
Dear Limma authors,
Thank you very much for this amazing package. In the recent use of the package, I found a potential issue when analyzing RNASeq data. After doing some investigation, I found the issue only happens using contrast.fit() when weight is included from voom()
To illustrate the issue, I use GSE137430 data: there are 3 time points (BL, W4 and W16) and 2 treatments (Secukinumab and Placebo) in the data set. I only used LS tissue type. Let's only focus on change from baseline by treatment
I generate grp = paste(Treatment, Timepoint, sep = "_"), and output the result for gene ID ENSG00000000003.14. Please note that all the results are before eBayes(). The issue is all the standard errors are a little bit different when the design matrix change or reference level of treatment change in the same design matrix (2 & 3 below), given all the coefficients/estimates are the same.
- design matrix - model.matrix(~0+grp, data = info1) contrasts.fit() output
And origSE is the standard error before eBayes()
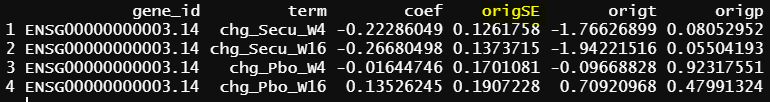
- design matrix - model.matrix(~1+Treatment*Timepoint, data=info1) treatment reference level is Placebo, and different design matrix from 1, but the model is practically the same. contrasts.fit() output
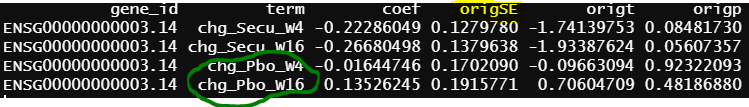
- design matrix - model.matrix(~1+Treatment*Timepoint, data=info2) treatment reference level is Secukinumab, the design matrix is the same as 2, the only different is Secukinumab is the reference level. contrasts.fit() output
As you can see all the coefficients across these three scenarios are the same, but SE and p values are slightly different.
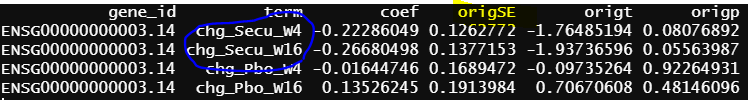
The reason I suspect the difference caused by contrast.fit() is that lmFit() output seems good for the common terms among the first two models
- design matrix - model.matrix(~0+grp, data = info1) lmFit() output
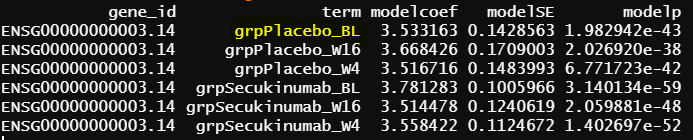
- design matrix - model.matrix(~1+Treatment*Timepoint, data=info1) placebo as reference lmFit() output
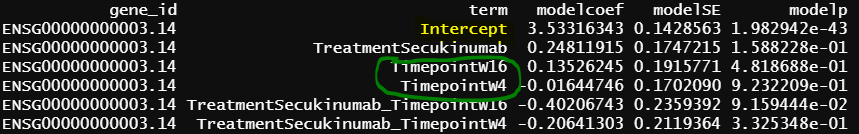
The intercept from model 2 is identical as grpPlacebo_BL output, and the intercept is the reference level of treatment and Timepoint (Placebo and Baseline). lmFit() output is probably correct.
Another interesting finding is the green circle part of the results are the same as the green circle part results from contrasts.fit(). The contrast for this output only has one term (detail code below), if lmFit() gives correct results, the result from contrasts.fit() with only one term is also correct, but issues happen when there more than one term.
- design matrix - model.matrix(~1+Treatment*Timepoint, data=info2) Secukinumab as reference lmFit() output
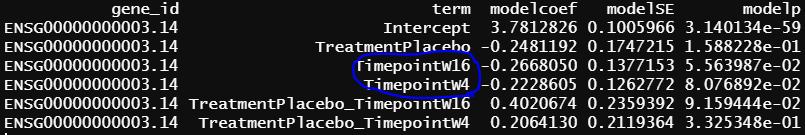
similar to previous model, blue circle info are the same as blue circle in contrasts.fit() output
The codes below show these example outputs. I did some further investigation and found that when the samples were independent, similar issues happened when weight is included. Without using weight, all the output of contrasts.fit() are identical among different model or different reference levels.
Please note that the difference of the p-values from contrasts.fit() are small - as you can see from the examples.
Happy to share the csv data sets used. Thank you very much.
Best, Zhenyu
library(data.table)
library(plyr)
library(tidyverse)
library(limma)
library(edgeR)
dir_in1 <- "C:/Users/wangz260/OneDrive - Pfizer/Downloads"
info0 <- fread(file.path(dir_in1, "GSE137430_Sample_Annotation.txt"))
info1 <- info0 %>% filter(tissuetype=="LS") %>%
mutate(grp = paste(Treatment, Timepoint, sep = "_"))
#Count table was generated by GSE137430 fastq files.
ct0 <- fread(file.path(dir_in1, "GSE137430_Gene-count-table.txt"))
ct1 <- ct0 %>% filter(gene_type=="protein_coding")
ct2 <- ct1 %>% select(-gene, -gene_name, -gene_type, -chr, -start, -end, -strand, -length)
rownames(ct2) = ct1$gene
ct3 <- as.matrix(ct2 %>% select(info1$Sample_ID))
#sanity check
colnames(ct3)==info1$Sample_ID
rownames(ct3) <- rownames(ct2)
dlist1 <- DGEList(counts=ct3)
#filter out low read genes
keep <- filterByExpr(dlist1, group = info1$grp, min.count=20, min.total.count=100)
sum(keep)
dlist2 <- dlist1[keep, , keep.lib.sizes=TRUE]
tmm1 <- calcNormFactors(dlist2,method="TMM")
desmat1 <- model.matrix(~0+grp, data = info1)
vres1 <- voom(tmm1, desmat1, plot=TRUE)
vcorfit1 <- duplicateCorrelation(vres1,desmat1,block=info1$Patient_ID)
#skip voomWithQualityWeights()
vfit1 <- lmFit(vres1, desmat1, block=info1$Patient_ID, correlation=vcorfit1$consensus, plot = TRUE)
vcont1 <- makeContrasts(
chg_Secu_W4 = grpSecukinumab_W4 - grpSecukinumab_BL,
chg_Secu_W16 = grpSecukinumab_W16 - grpSecukinumab_BL,
chg_Pbo_W4 = grpPlacebo_W4 - grpPlacebo_BL,
chg_Pbo_W16 = grpPlacebo_W16 - grpPlacebo_BL,
levels = desmat1)
vfit1cont1 <- contrasts.fit(vfit1, vcont1)
# get the lmFit() model output
modelcoef1 <- vfit1$coef
modelcoef2 <- modelcoef1%>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelcoef")
modelSE1 <- vfit1$stdev.unscaled * vfit1$sigma
modelSE2 <- modelSE1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelSE")
modelt1 <- vfit1$coef / vfit1$stdev.unscaled / vfit1$sigma
modelt2 <- modelt1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelt")
modelp1 <- 2 * pt(-abs(modelt1), df = vfit1$df.residual)
modelp2 <- modelp1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelp")
modelcoef3 <- modelcoef2 %>% left_join(modelSE2) %>%left_join(modelp2)
# get contrasts.fit() output before eBays()
SE1 <- vfit1cont1$stdev.unscaled * vfit1cont1$sigma
SE2 <- SE1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origSE")
t1 <- vfit1cont1$coef / vfit1cont1$stdev.unscaled / vfit1cont1$sigma
t2 <- t1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origt")
p1 <- 2 * pt(-abs(t1), df = vfit1cont1$df.residual)
p2 <- p1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origp")
coef1 <- vfit1cont1$coef
coef2 <- coef1%>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "coef")
coef3 <- coef2 %>% full_join(SE2) %>% left_join(t2) %>% left_join(p2)
#save the info
inter_0_model <- modelcoef3
inter_0_contrast <- coef3
#####
#in order to show the issue, a different design matrix is needed.
#For the current info1, the reference level of treatment is Placebo
#####
desmat2 <- model.matrix(~1+Treatment*Timepoint, data=info1)
colnames(desmat2) <- gsub(":", "_", colnames(desmat2))
colnames(desmat2) <- gsub("\\(|\\)", "", colnames(desmat2))
vfit2 <- lmFit(vres1, desmat2, block=info1$Patient_ID, correlation=vcorfit1$consensus, plot = TRUE)
vcont2 <- makeContrasts(
chg_Secu_W4 = TreatmentSecukinumab_TimepointW4 + TimepointW4,
chg_Secu_W16 = TreatmentSecukinumab_TimepointW16 + TimepointW16,
chg_Pbo_W4 = TimepointW4,
chg_Pbo_W16 = TimepointW16,
levels = desmat2)
vfit2cont1 <- contrasts.fit(vfit2, vcont2)
# get the lmFit() model output
modelcoef1 <- vfit2$coef
modelcoef2 <- modelcoef1%>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelcoef")
modelSE1 <- vfit2$stdev.unscaled * vfit2$sigma
modelSE2 <- modelSE1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelSE")
modelt1 <- vfit2$coef / vfit2$stdev.unscaled / vfit2$sigma
modelt2 <- modelt1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelt")
modelp1 <- 2 * pt(-abs(modelt1), df = vfit2$df.residual)
modelp2 <- modelp1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelp")
modelcoef3 <- modelcoef2 %>% left_join(modelSE2) %>%left_join(modelp2)
# get contrasts.fit() output before eBays()
SE1 <- vfit2cont1$stdev.unscaled * vfit2cont1$sigma
SE2 <- SE1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origSE")
t1 <- vfit2cont1$coef / vfit2cont1$stdev.unscaled / vfit2cont1$sigma
t2 <- t1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origt")
p1 <- 2 * pt(-abs(t1), df = vfit2cont1$df.residual)
p2 <- p1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origp")
coef1 <- vfit2cont1$coef
coef2 <- coef1%>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "coef")
coef3 <- coef2 %>% full_join(SE2) %>% left_join(t2) %>% left_join(p2)
#save the info
ref_pbo_model <- modelcoef3
ref_pbo_contrast <- coef3
#####
#Change the reference level of treatment to Secukinumab
#####
info2 <- info1 %>% mutate(Treatment=factor(Treatment, levels = c("Secukinumab", "Placebo")))
desmat3 <- model.matrix(~1+Treatment*Timepoint, data=info2)
colnames(desmat3) <- gsub(":", "_", colnames(desmat3))
colnames(desmat3) <- gsub("\\(|\\)", "", colnames(desmat3))
vfit3 <- lmFit(vres1, desmat3, block=info2$Patient_ID, correlation=vcorfit1$consensus, plot = TRUE)
vcont3 <- makeContrasts(
chg_Secu_W4 = TimepointW4,
chg_Secu_W16 = TimepointW16,
chg_Pbo_W4 = TreatmentPlacebo_TimepointW4 + TimepointW4,
chg_Pbo_W16 = TreatmentPlacebo_TimepointW16 + TimepointW16,
levels = desmat3)
vfit3cont1 <- contrasts.fit(vfit3, vcont3)
# get the lmFit() model output
modelcoef1 <- vfit3$coef
modelcoef2 <- modelcoef1%>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelcoef")
modelSE1 <- vfit3$stdev.unscaled * vfit3$sigma
modelSE2 <- modelSE1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelSE")
modelt1 <- vfit3$coef / vfit3$stdev.unscaled / vfit3$sigma
modelt2 <- modelt1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelt")
modelp1 <- 2 * pt(-abs(modelt1), df = vfit3$df.residual)
modelp2 <- modelp1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "modelp")
modelcoef3 <- modelcoef2 %>% left_join(modelSE2) %>%left_join(modelp2)
# get contrasts.fit() output before eBays()
SE1 <- vfit3cont1$stdev.unscaled * vfit3cont1$sigma
SE2 <- SE1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origSE")
t1 <- vfit3cont1$coef / vfit3cont1$stdev.unscaled / vfit3cont1$sigma
t2 <- t1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origt")
p1 <- 2 * pt(-abs(t1), df = vfit3cont1$df.residual)
p2 <- p1 %>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "origp")
coef1 <- vfit3cont1$coef
coef2 <- coef1%>% data.frame %>% mutate(gene_id = rownames(.)) %>%
pivot_longer(., -gene_id, names_to = "term", values_to = "coef")
coef3 <- coef2 %>% full_join(SE2) %>% left_join(t2) %>% left_join(p2)
#save the info
ref_secu_model <- modelcoef3
ref_secu_contrast <- coef3
#####
#Compare different model output, use gene ID ENSG00000000003.14 as example
#####
inter_0_contrast %>% filter(gene_id=="ENSG00000000003.14") %>% data.frame
ref_pbo_contrast %>% filter(gene_id=="ENSG00000000003.14") %>% data.frame
ref_secu_contrast %>% filter(gene_id=="ENSG00000000003.14") %>% data.frame
inter_0_model %>% filter(gene_id=="ENSG00000000003.14") %>% data.frame
ref_pbo_model %>% filter(gene_id=="ENSG00000000003.14") %>% data.frame
ref_secu_model %>% filter(gene_id=="ENSG00000000003.14") %>% data.frame
sessionInfo( )
R version 4.0.2 (2020-06-22)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19044)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.1252 LC_CTYPE=English_United States.1252 LC_MONETARY=English_United States.1252
[4] LC_NUMERIC=C LC_TIME=English_United States.1252
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] edgeR_3.32.1 limma_3.52.2 forcats_0.5.0 stringr_1.4.0 dplyr_1.0.2 purrr_0.3.4 readr_1.3.1
[8] tidyr_1.1.2 tibble_3.0.3 ggplot2_3.3.2 tidyverse_1.3.0 plyr_1.8.6 data.table_1.14.0
loaded via a namespace (and not attached):
[1] statmod_1.4.34 tinytex_0.26 locfit_1.5-9.4 tidyselect_1.1.0 xfun_0.17 splines_4.0.2
[7] haven_2.3.1 lattice_0.20-41 colorspace_1.4-1 vctrs_0.3.8 generics_0.0.2 utf8_1.1.4
[13] blob_1.2.1 rlang_0.4.11 pillar_1.4.6 glue_1.4.2 withr_2.4.3 DBI_1.1.0
[19] bit64_4.0.5 BiocGenerics_0.36.0 dbplyr_1.4.4 modelr_0.1.8 readxl_1.3.1 lifecycle_1.0.1
[25] munsell_0.5.0 gtable_0.3.0 cellranger_1.1.0 rvest_0.3.6 Biobase_2.50.0 parallel_4.0.2
[31] fansi_0.4.1 broom_0.7.4 Rcpp_1.0.5 backports_1.1.10 scales_1.1.1 jsonlite_1.7.1
[37] bit_4.0.4 fs_1.5.0 hms_0.5.3 stringi_1.5.3 grid_4.0.2 cli_3.1.0
[43] tools_4.0.2 magrittr_1.5 crayon_1.3.4 pkgconfig_2.0.3 ellipsis_0.3.1 Matrix_1.2-18
[49] xml2_1.3.2 NOISeq_2.34.0 reprex_0.3.0 lubridate_1.7.9 assertthat_0.2.1 httr_1.4.2
[55] rstudioapi_0.13 R6_2.4.1 compiler_4.0.2

Thank you for the response, Gordon. I have a followup question: If model.matrix(~0+grp, data = info1) gives exact standard error from contrasts.fit(), should we expect the contrasts.fit() results (first screenshot) are the same as lmFit() outputs in green circle and blue circles (last two screenshot from the OP)? I think lmFit() gives exact standard error. Thank you.
The results from lmFit() are always exact regardless of design matrix or weights.
Sorry I cannot comment on your green and blue circles. The output you show is not standard limma output and I don't have time to go through your complex code to figure out what the screenshots represent.
Hi Gordon, Sorry for the complex code. I wanted to output the results of contrasts.fit() before eBayes() so that we can compare lmFit() output with contrasts.fit()'s. The code was to get the coefficient, SE, test statistics and p-value from the contrasts.fit() (before eBays()). If there's a easier way to get those info, please let me know and I'll rewrite my code. Thank you very much.
P.S. I was able to replicate the results (yellow and blue circles) using nlme::gls() and lsmeans() and contrast() from emmean package. with the consensus correlation and weights from voom().
There is no reason for doing what you are doing for any real data analysis so it is outside the scope of what I provide advice on. If you want to check whether the
contrasts.fitoutput is exact, you only need to look atfit$stdev.unscaled.lmFithas always been exact so I'm not sure why you are trying to replicate it using other packages.Thank you, Gordon. I used
fit$stdev.unscaled * fit$sigmato check the contrasts.fit SE output, and that's when I found the SE discrepancies (contrasts.fit() from model.matrix(~0+grp) vs lmFit() output from model.matrix(~1+Treatment*Timepoint)). And that's why I used other packages to see which results I can replicate. If you have more free time, it'll be great if you can take another look at my OP. Thanks again for providing the information.I have no free time, but I still do my best to help people if I can.
I have already answered your questions, and the answers are in the limma documentation anyway. If you use
~0+grp(and there are no blocks shared between the groups) thencontrasts.fitmust agree exactly withlmFitbecause both are exact. Otherwise they won't be. What else do you need to know? If you want more help then please:Hi Gordon, thank you again for taking the time to reply my post. I'll start a new post with your suggestions. I found that contrasts.fit() results from model.matrix(~0+grp) is not exact and they are different from lmFit() output, and I tried to demonstrate that. In order to show you the comparison, I have to show the result before the eBayes().
OK, I notice that you are using duplicateCorrelation and blocks. The text of your question didn't mention using blocks but I see the use of duplicateCorrelation in the code.
The reason why contrasts.fit doesn't agree exactly with lmFit in your case, even with the group-means parametrization, will presumably be because there are blocks shared between the groups. (I can't confirm because you haven't shown your data but it must be so.) That will cause the group means being compared to be not independent and hence to contrasts.fit not being exact.
I have now added a new answer below to show you in more detail when contrasts.fit will be exact and when it will not.