
Hello, I would like to confirm if the low assignment ratio (54%) is normal, and please check the possible reason I found.
I used Hisat2 to assign paired-end strand-specific transcriptomic sequences (rRNA removed) to a reference genome. Because I filtered out the unmapped sequences in advance, the overall assignment ratio displayed by Hisat2 was 100%, and the multi-mapping ratio was only 0.3%. The following is the Hisat2 command.
hisat2 -x reference/index -1 mRNA_paired_fwd.fq.gz -2 mRNA_paired_rev.fq.gz -S paired.sam --very-sensitive --fr -t -p 10 --rna-strandness RF --no-spliced-alignment --no-softclip
Then I wanted to use FeatureCounts to get read counts based on the bam file obtained by Hisat2, below is my command, but successful assignment ratio was only about 54%, the attribute of others was "Unassigned_NoFeatures". It was confusing, the GFF file was obtained via Prodigal, it should only be Including CDS, and there is no sequence such as rRNA. So, for featureCounts, there should be a relatively high assignment ratio.
featureCounts -t CDS -g ID -a reference.gff -o counts.txt paired.bam -s 2 -T 64 -M --primary -p -F GFF
Then, I added "-R" to see the unassigned transcriptomic sequences. For convenience, we picked an unassigned sequence, and named it unassigned.fa, which was 150 nt. We found that the unassigned.fa overlapped with the reference genome (region start-end: 3830-3681), and the GFF file showed that the overlapped region (region start-end: 2797-3840) was a CDS. Therefore, the unassigned.fa should be successfully assigned, but it was not. Meanwhile, some sequnces that overlapped the same region (region start-end: 2797-3840) were counted.
Then I viewed the bam file, and found that only the sequences whose bam flags were 163 and 83 were assigned to features. Therefore, I think that the low assignment ratio was because the stranded-specific libraries were constructed by dUTP method.
However, the assignment ratio of some samples was only 2~3%, and the libraries were also constructed by dUTP method.
Could you please check this possible reason?
Please let me know if additional information is required.
Mort
Thanks in advance!
Your tags are already good but I've added an Rsubread tag, which will make absolutely sure that the Rsubread developers see your post. Be aware though that Australia has a 5-day break for Easter.
Thanks for the reminder of the holiday.
Apologies for the impolite and inappropriate reply in other posts!
What organism is this? If this is mouse data, then from my experiences the percentage of assigned reads is typically around 50-70%, so your percentage is within the range. Regarding the example read you showed, how did you obtain its mapping location? Was it the one reported in the bam file? Lastly, regarding some of your samples having only 2-3% of reads assigned, I am not sure what is the reason. You may try to set '-s 1', or there may be an issue with the sample prep for these samples.
Thanks for your answers!
The metatranscriptomes were from mixed uncultured microbiota. The percentage of assigned reads in most samples is around 50-70%.
For the mapping location of the unassigned sequence, I used the following command to get the mapping location.
nucmer --maxmatch --nosimplify unassigned.fa reference_genome.fashow-coords -lrcTH out.deltaYes, the unassigned sequence was reported in the bam file. The bam flags of all assigned sequences with features are 163 or 83. The 163 flag represents that read paired (0x1), read mapped in proper pair (0x2), mate reverse strand (0x20) and second in pair (0x80). The 83 flag represents that read paired (0x1), read mapped in proper pair (0x2), read reverse strand (0x10) and first in pair (0x40). The bam flags of all sequences with nofeature are 147 or 99. The 147 flag represents that read paired (0x1), read mapped in proper pair (0x2), read reverse strand (0x10) and second in pair (0x80). The 99 flag represents that read paired (0x1), read mapped in proper pair (0x2), mate reverse strand (0x20) and first in pair (0x40). The metatranscriptomics libraries were constructed using dUTP method, shoud this be a possible reason?
Thanks in advance!
You need to use the mapping locations reported in your bam file to cross check the featureCounts counting results, as your HiSat mapping result for your example reads might be different from your nucmer command. The dUTP protocol is commonly used for performing strand-specific sequencing. You may try to carry out a unstranded read counting (use default value of -s) to see if you can get a reasonable counting percentage. If you still couldn't then that will indicate that there might be issues with those few samples.
Thanks for your prompt reply.
Please check the following information of bam file and gff file.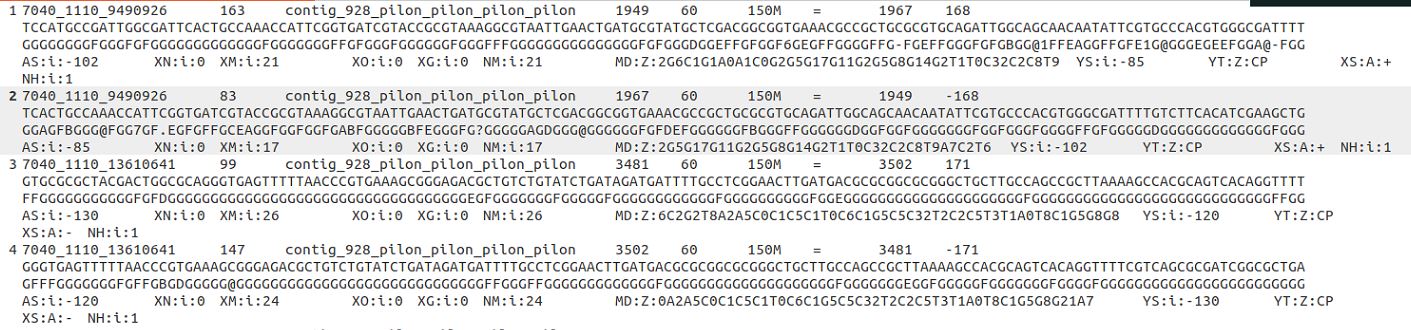
The paired reads 7040_1110_9490926 are assigned to the feature 1-2, the read length is 150 nt.
According to the mapping locations of the paired reads 7040_1110_13610641, the paired reads 7040_1110_13610641 should be assigned to feature 1-3, but it was labeled with Unassigned_NoFeatures. Could you please check this?
Sorry for the wrong description in the previous post. When setting '-s' as 1, the percentage of assigned reads is decreased significantly.
When using a unstranded read counting, the percentage of assigned reads is increased significantly, and the paired reads 7040_1110_13610641 are assigned to feature 1-3. The result is reasonable. So, should I use unstranded read counting method to re-generate count files, even if the libraries were constructed by dUTP method, for example the following command.
Thanks in advance!
Thanks for giving very detailed read mapping results and counting results. I assume that the issue that you reported (i.e. 7040_1110_9490926 was assigned to features 1_2, while 7040_1110_13610641 was reported as "no features" ) was observed in the result of featureCounts run with
-s 2. This is because you did pair-end read assignment. In the most common settings of paired-end read sequencing with Illumina sequencers, the second read is from the opposite strand of the whole fragment. Say, a whole fragment is from the positive strand of the chromosome, then R1 will have the sequence of the positive strand of that chromosome, but R2 will have the sequence of the negative strand of the chromosome. Hence, the read aligner always maps R1 and R2 to different strands if the read-pair contains no errors and the aligner did correct read mapping.By knowing this, featureCounts always flips the "strandness" of the read with the 0x80 flag (i.e. R2) to its opposite strand. Because the "R2" read in 7040_1110_13610641 (i.e. the read with flag 147) was mapped to the negative strand, featureCounts flipped the strandness of R2, suggesting that the whole fragment was from the positive strand of the chromosome. This is consistent with the strandness of "R1" in the same read-pair: R1 has a flag of 99; this flag also suggests that the whole fragment was from the positive strand of the chromosome.
Then, when featureCounts was run with
-s 2, it only searched for the features that are on the opposite strand of the whole fragment of 7040_1110_13610641; that is to say, featureCounts only tried to find the annotations on the negative strand (-) for assigning this read-pair. On the other hand, all the features listed in your screenshot are from the positive strand (+). Hence featureCounts found no features for read-pair 7040_1110_13610641.The other read-pair in your screenshot, 7040_1110_9490926, has R1 mapped to the negative strand (flag = 83) and R2 mapped to the positive strand of the chromosome (flag = 163). Hence featureCounts takes it as the whole fragment of 7040_1110_9490926 was from the negative strand of the chromosome. When featureCounts was run with
-s 2, it only searched for features from the positive strand in the annotations for this read-pair, hence you had this read-pair assigned to feature 1_2.Thank you very much for your clear answer!
So, is it feasible to set '-s 0' for strand-specific transcriptomics? Because I think there is indeed a sequence alignment to this region.
Thanks in advance!
If your data was indeed generated from strand-specific sequencing, then performing strand-specific counting should give you better results. There seems to be issues with your data to me because both modes of strand-specific counting did not produce reasonable results but unstranded counting produced reasonable results. You may need to talk to your sequencing facility to see if the samples of concern were indeed generated from strand-specific sequencing.
Thank you very much for your detailed and patient answer!
Thank you very much for your detailed and patient answer!