Entering edit mode

I am constructing phylogenetic tree as written in Bioconductor Workflow for Microbiome Data Analysis: from raw reads to community analyses [version 2; peer review: 3 approved]. I have 242 samples that contain 41684 ASVs. The code:
dm <- dist.ml(phang.align)
resulted in data object (dm) of 13.9 GB (sic!), the next line cannot be performed due to memory shortage
> treeNJ <- NJ(dm)
Error: vector memory exhausted (limit reached?)
is there any other way to complete the code part? Are there any other solutions available? like splitting the 41k ASVs into batches, making several small trees and later join the trees?
> sessionInfo()
R version 4.0.3 (2020-10-10)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur 10.16
Matrix products: default
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] phangorn_2.6.3 ape_5.5
loaded via a namespace (and not attached):
[1] nlme_3.1-152 bitops_1.0-7 matrixStats_0.58.0 phyloseq_1.34.0
[5] bit64_4.0.5 RColorBrewer_1.1-2 progress_1.2.2 httr_1.4.2
[9] GenomeInfoDb_1.26.7 tools_4.0.3 vegan_2.5-7 utf8_1.2.1
[13] R6_2.5.0 mgcv_1.8-35 DBI_1.1.1 BiocGenerics_0.36.1
[17] colorspace_2.0-0 permute_0.9-5 rhdf5filters_1.2.0 ade4_1.7-16
[21] tidyselect_1.1.0 prettyunits_1.1.1 DESeq2_1.30.1 bit_4.0.4
[25] compiler_4.0.3 microbiome_1.13.8 Biobase_2.50.0 DelayedArray_0.16.3
[29] scales_1.1.1 quadprog_1.5-8 genefilter_1.72.1 stringr_1.4.0
[33] Rsamtools_2.6.0 dada2_1.18.0 XVector_0.30.0 jpeg_0.1-8.1
[37] pkgconfig_2.0.3 MatrixGenerics_1.2.1 fastmap_1.1.0 rlang_0.4.10
[41] rstudioapi_0.13 RSQLite_2.2.7 generics_0.1.0 hwriter_1.3.2
[45] jsonlite_1.7.2 BiocParallel_1.24.1 dplyr_1.0.5 RCurl_1.98-1.3
[49] magrittr_2.0.1 GenomeInfoDbData_1.2.4 biomformat_1.18.0 Matrix_1.3-2
[53] Rhdf5lib_1.12.1 Rcpp_1.0.6 munsell_0.5.0 S4Vectors_0.28.1
[57] fansi_0.4.2 lifecycle_1.0.0 stringi_1.5.3 MASS_7.3-53.1
[61] SummarizedExperiment_1.20.0 zlibbioc_1.36.0 rhdf5_2.34.0 Rtsne_0.15
[65] plyr_1.8.6 grid_4.0.3 blob_1.2.1 parallel_4.0.3
[69] crayon_1.4.1 lattice_0.20-41 Biostrings_2.58.0 splines_4.0.3
[73] multtest_2.46.0 annotate_1.68.0 hms_1.0.0 locfit_1.5-9.4
[77] pillar_1.6.0 igraph_1.2.6 GenomicRanges_1.42.0 geneplotter_1.68.0
[81] reshape2_1.4.4 codetools_0.2-18 stats4_4.0.3 fastmatch_1.1-0
[85] XML_3.99-0.6 glue_1.4.2 ShortRead_1.48.0 latticeExtra_0.6-29
[89] data.table_1.14.0 RcppParallel_5.1.2 png_0.1-7 vctrs_0.3.7
[93] foreach_1.5.1 tidyr_1.1.3 gtable_0.3.0 purrr_0.3.4
[97] assertthat_0.2.1 cachem_1.0.4 ggplot2_3.3.3 xfun_0.22
[101] xtable_1.8-4 survival_3.2-11 tibble_3.1.1 iterators_1.0.13
[105] GenomicAlignments_1.26.0 tinytex_0.31 AnnotationDbi_1.52.0 memoise_2.0.0
[109] IRanges_2.24.1 cluster_2.1.2 ellipsis_0.3.1


Hi Axel! Thanks for your comment, that's something new for me! I do not know what to expect. I thought the higher number the better. I just switched from OTUs. I have human stool microbiome samples, V3-V4 amplicons sequenced with 2x300PE. My QC of R1 and R2 reads are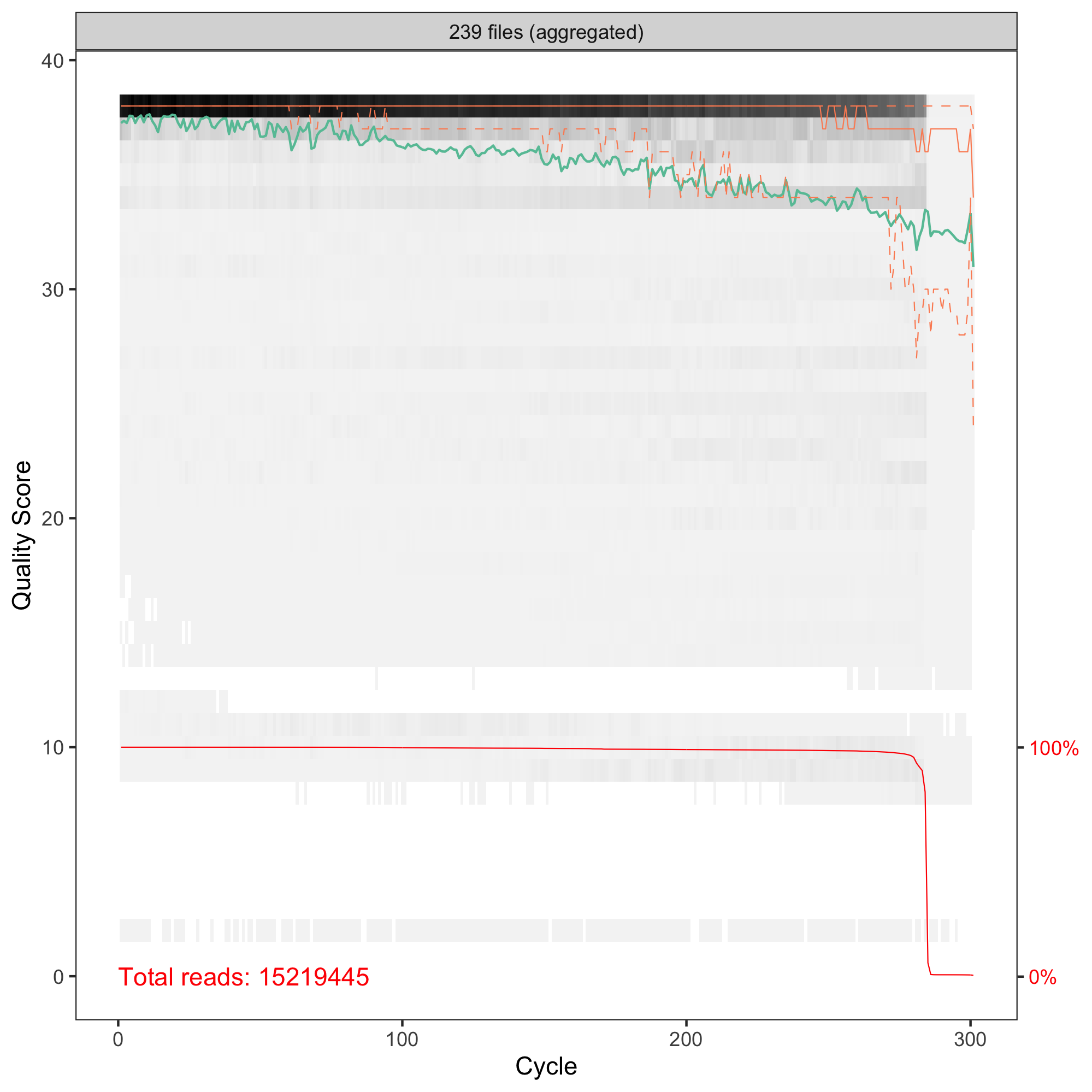 and
and 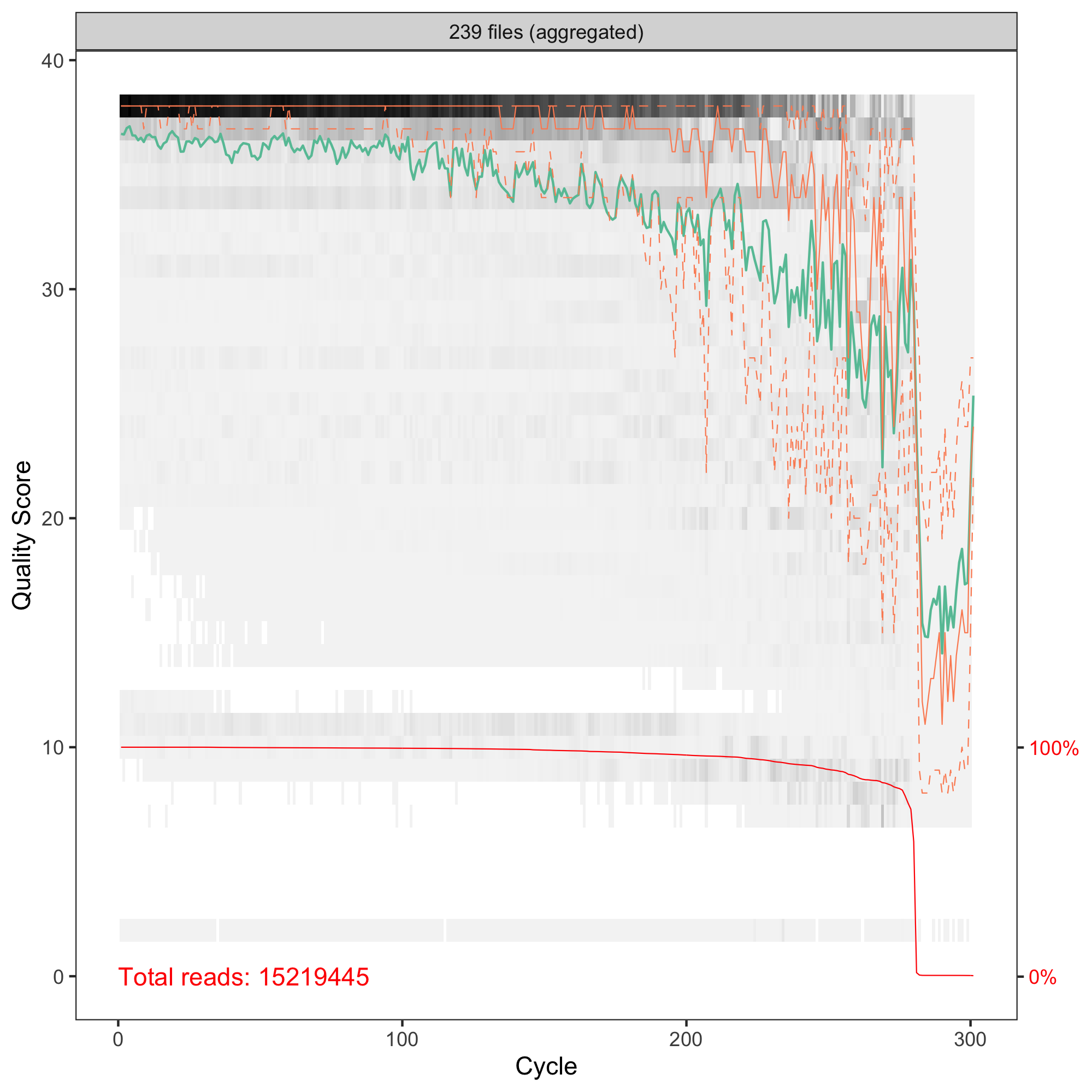 so I set
so I set 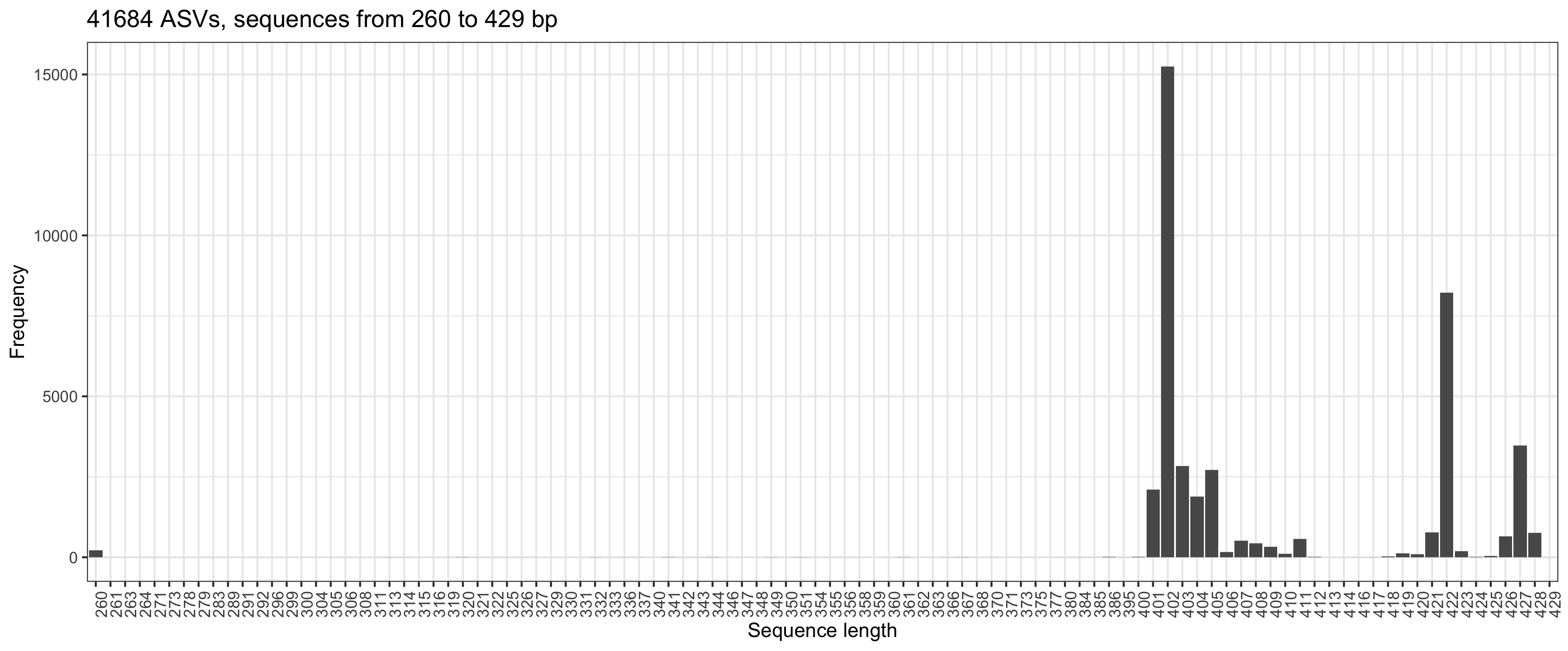 . Can you comment and give me some advice. Best regards, Marcin
. Can you comment and give me some advice. Best regards, Marcin
truncLen = c(260, 181)in thefilterAndTrim. Longer sequences resulted in a lower number of ASVs (but still > 36K). Merging R1 and R2 gave sequences from 260 to 429 bpHi Marcin, ok, human stool is not that exceptional. :-)
I have realized in the meantime that the filtering I thought of is actually done after constructing the phylogenetic tree in the workflow, in the phyloseq section on pages 10-12. I would think that postponing the tree construction is an option in your case although I'm not an expert here.
If you have set truncLen to c(260, 181), then the two groups to the right of your plot (at 422 and 427) will have an overlap of 10-20 bases only and I'd be inclined to remove them as well as the funny ones at 260.
Have you used any of the other arguments to filterAndTrim()? The published workflow is using maxEE = 2 and I have experimented with this one quite a bit.
Usually I aim at removing 10%-20% of the reads during QC and retaining about 1000 ASVs after filtering.
Best,
Hi Axel, I am using the settings from DADA2 Pipeline Tutorial. There are maxN=0, maxEE=c(2,2), truncQ=2, and rm.phix=TRUE. Removing the small ones and these >422 seems a good idea. Does lowering the maxEE and truncQ will make the selection more stringent? Best regards, Marcin
Hi Marcin, yes, lowering these threshold should result in more stringent quality filtering, although you don't have many choices with a default of 2. And of course, how much more stringent will depend on your data. In our in-house example I have checked yesterday, I actually had to raise the threshold to avoid losing more than 50% of the reads (human saliva), especially of the reverse reads.
If that doesn't bring the number of ASVs down far enough, it may help to look at the counts per ASV and sample, and filter ASVs based on minimum counts or mininmum occurrence in samples, depending on your research question.
Different tree-building algorithms may have different memory requirements but I cannot advise on this.
Best regards,
Thanks again! I have tried longer sequences
truncLen = c(265, 185)to allow more overlap and the number of ASVs dropped to 1368, and afterremoveBimeraDenovoto 1058. That should be good right now. Best regards, Marcin1058 sounds much, much better. :-) Good to hear you got it done.
Cheers,