
As I wanted to retrieve all relevant genetic annotation for human genome using AnnotationDbi package as well as reference database org.Hs.eg.db, I have noticed following seemingly odd observation.
NCBI_gene_annotationDbi <- AnnotationDbi::select(org.Hs.eg.db, keys=AnnotationDbi::keys(org.Hs.eg.db, keytype="SYMBOL"), columns=c("SYMBOL", "GENENAME","ENTREZID"), keytype="SYMBOL")
duplicated_genes <- NCBI_gene_annotationDbi$SYMBOL[duplicated(NCBI_gene_annotationDbi$SYMBOL)]
NCBI_gene_annotationDbi_duplicated <- NCBI_gene_annotationDbi %>% dplyr::filter(SYMBOL %in% duplicated_genes)
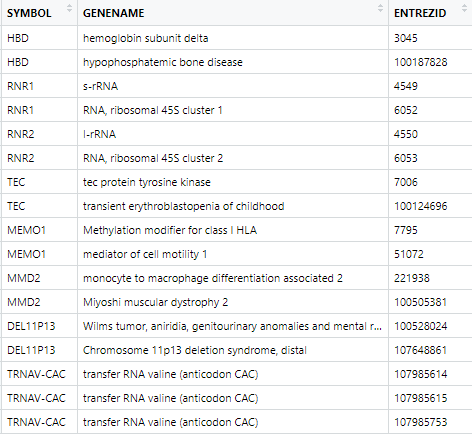
As you can notice, only a small fraction (8 symbol over a total 61538) of SYMBOL have duplicated inputs in ENTREZID field. Besides, most of these duplicated values often integrate a general description and annotation of the gene, while the other ones seem superfluous (for instance, HBD has two ENTREZID matches, one general about hemoglobin subunit deltat, and another one really specific, and not describing its function: bone disease).
For such a small number of duplicated inputs, can't it be possible to clear all these seemingly superfluous one-to-many matches, only keeping for each the most relevant one. Or maybe merge both descriptions?
sessionInfo( )
R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /softhpc/R/4.0.2/lib64/R/lib/libRblas.so
LAPACK: /softhpc/R/4.0.2/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] GO.db_3.12.1 hgu133plus2probe_2.18.0 hgu133plus2.db_3.2.3 org.Hs.eg.db_3.12.0
[5] hgu133plus2cdf_2.18.0 bmkanalysis_1.0.0 testthat_3.0.1 affy_1.68.0
[9] EnsDb.Hsapiens.v86_2.99.0 ensembldb_2.14.0 AnnotationFilter_1.14.0 GenomicFeatures_1.42.1
[13] AnnotationDbi_1.52.0 Biobase_2.50.0 GenomicRanges_1.42.0 GenomeInfoDb_1.26.2
[17] IRanges_2.24.1 S4Vectors_0.28.1 BiocGenerics_0.36.0


I see what you mean. However, only a tiny fraction of all SYMBOL genes match more than one ENTREZID input (precisely 8 symbol over a total 61538, with 17 cases to deal with). Seeing that, I would be highly tempted to consider either some of descriptions are redundant, or as you suggest, that they do describe different biological units, and as a result, should have their own unique SYMBOL input.
Taking HBD example, I have then checked on TxDb.Hsapiens.UCSC.hg19.knownGene database, and EnsDb.Hsapiens.v86. Conclusion: only one gene was reported, the one bearing ENTREZID 3045. In that particular case, hyphophosphatemic bone disease input seems to be erroneous, superfluous, or maybe really specific to a biological condition (seems odd to only describe a gene by the disease it triggers, doesn't it)?
Anyway, do you know which channel I could use to report directly NCBI from that possible mistake/redundancy?
The latter is a GeneRIF, which is something I had no idea existed until like five minutes ago. It's intended to allow people to add functional annotation to genes? But somehow there doesn't seem to be a gene involved for the GeneRIF HBP. But there do seem to be lots of these things. And the examples they provide appear to be real, like you know, genes.
So, I don't know. You could probably talk to somebody at NCBI to see what's up. I don't have any insight as to whomever that might be, so if you really care it's on you to find out.
Thanks for trying to enlighten me on the subject, and if I get newer information, I will come back to you.