Entering edit mode

vi.hammer
•
0
@user-24201
Last seen 4.3 years ago
I am currently comparing methylation calls from deepsignal and nanopolish using DSS. Using the showOneDMR function, this plot is generated:
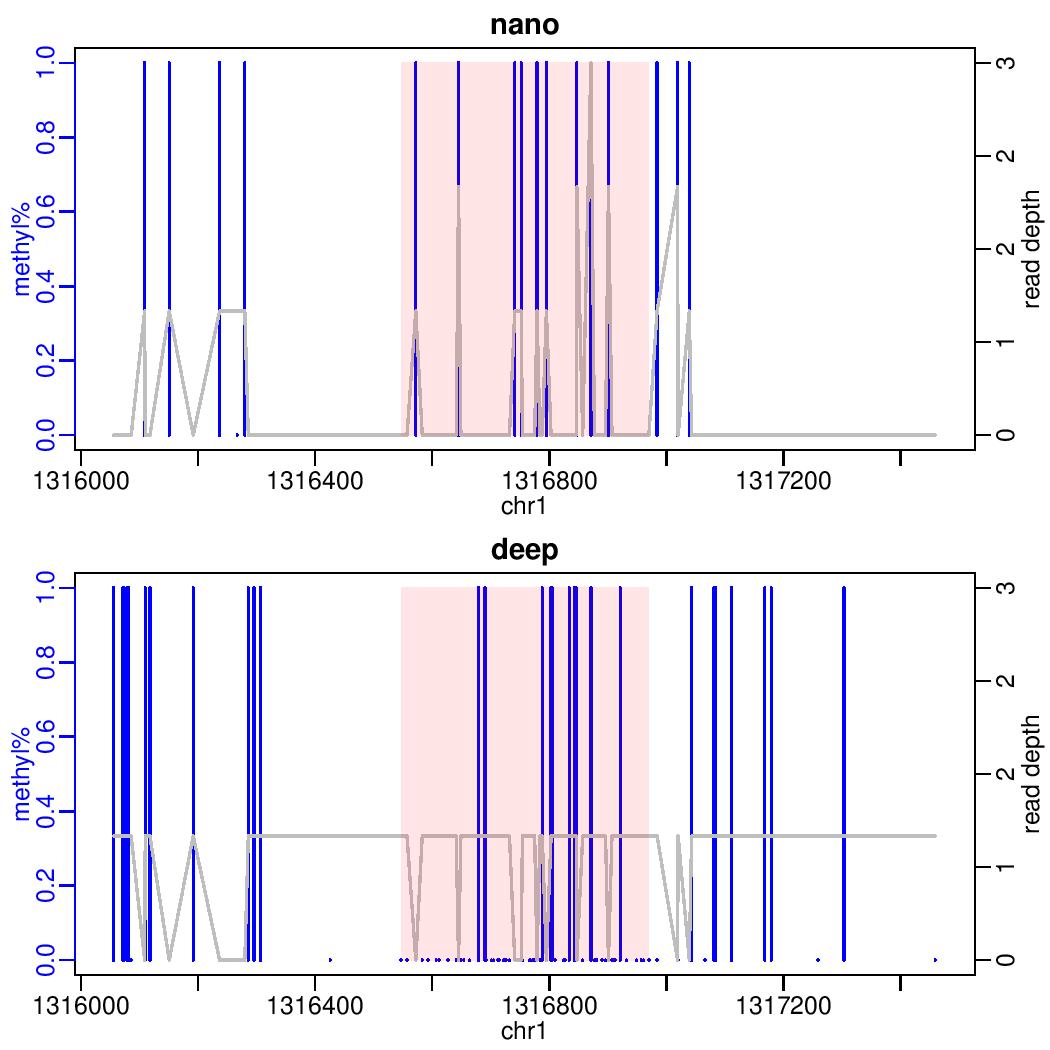
Note the axis labels for the read depth.
My R script:
library(DSS)
nano = read.table(file = 'nanopolish_methylation_freq.tsv', sep = '\t', header = TRUE)
deep = read.table(file = 'deepsignal_fast5s_single.freq-perCG-raw.tsv', sep = '\t', header = FALSE)
colnames(deep) <- c('chromosome','pos','strand','0_based_pos',
'prob_unmethy_sum','prob_methyl_sum',
'count_modified','count_unmodified',
'coverage','mod_freq','k_mer')
keeps <- c("chromosome", "start", "called_sites", "called_sites_methylated")
nano = nano[keeps]
keeps <- c("chromosome", "0_based_pos", "coverage", "count_modified")
deep = deep[keeps]
names(nano)[names(nano) == "start"] <- "pos"
names(nano)[names(nano) == "chromosome"] <- "chr"
names(nano)[names(nano) == "called_sites"] <- "N"
names(nano)[names(nano) == "called_sites_methylated"] <- "X"
names(deep)[names(deep) == "0_based_pos"] <- "pos"
names(deep)[names(deep) == "chromosome"] <- "chr"
names(deep)[names(deep) == "coverage"] <- "N"
names(deep)[names(deep) == "count_modified"] <- "X"
BSobj = makeBSseqData(list(nano, deep),
c("nano", "deep"))[1:10000]
dmlTest = DMLtest(BSobj, group1 = c("nano"),group2 = c("deep"), smoothing = TRUE)
dmrs = callDMR(dmlTest, p.threshold = 0.01)
showOneDMR(dmrs[1,], BSobj)

Using the updated version: https://github.com/haowulab/DSS/blob/master/R/BSseq_plots.R fixes the bug. Thank you!