Entering edit mode

Ricardo
•
0
@ricardo-24247
Last seen 4.3 years ago
Dear Methylomics community,
Using standard ChAMP 2.20.1 package to perform differential methylation analysis, we found very very different results if inputs are A vs B or if they are B vs A, where A and B are sets of properly pre-processed patient M-values. We expected the same (symmetrical) results (those up- in A-vs-B comparison being down- in B-vs-A and so on) but this did not happen. If more experienced colleagues could shed some light on this issue we would greatly appreciate.
MVals_Q <- read.table("../Dados.RVencio/MVals_Q.csv", sep = "\t", header = TRUE)
targets <- read.table("../Dados.RVencio/targets.csv", sep = "\t", header = TRUE)
Values <- dplyr::select(as.data.frame(MVals_Q), matches(c("A|B")))
names(Values) <- gsub ("A_","",names(Values))
names(Values) <- gsub ("B_","",names(Values))
aux1 <- subset(targets, tagets$Sample_Group == "A" | targets$Sample_Group =="B" )
aux1$Classification[aux1$Sample_Group == "A"] <- "A"
aux1$Classification[aux1$Sample_Group == "B"] <- "B"
targets.m <- aux1
myDMP <- champ.DMP(beta = Values,
pheno=targets.m$Classification,
adjPVal = 0.01,
compare.group=NULL,
adjust.method = "BH",
arraytype = "EPIC")
The screen print is in the following:
A-vs-B
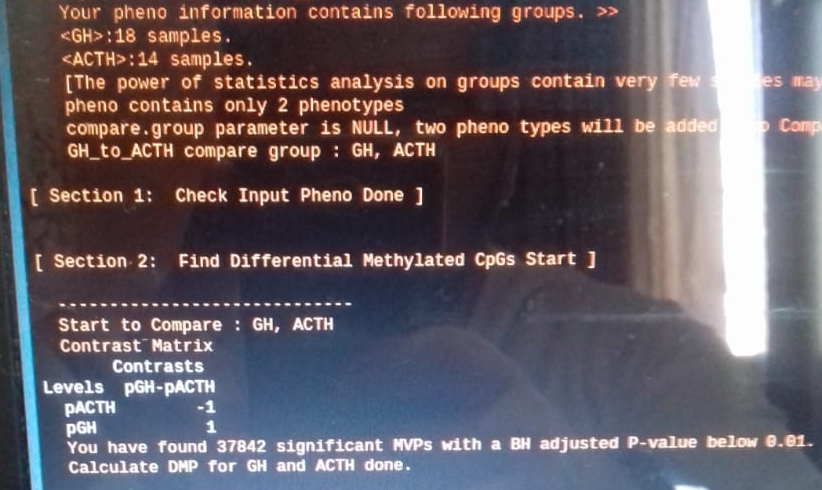
and B-vs-A, the same datset and R-code
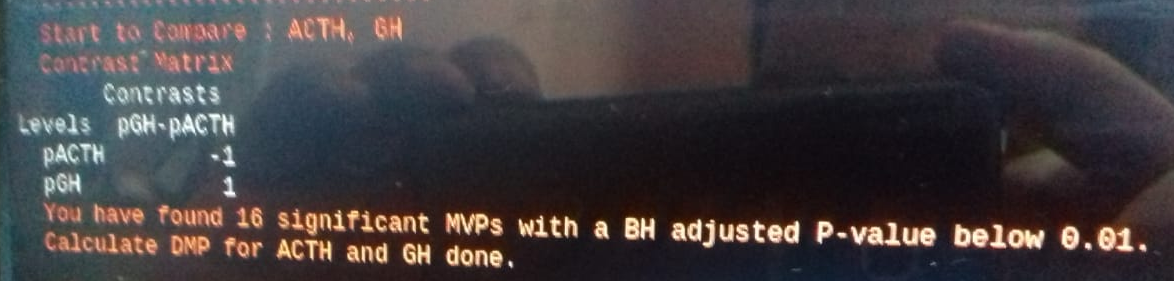
Thank you very much in advance for your time
Ricardo Vêncio and Junier Marrero
