
Hi,
I have a question regarding large differences in library size amongst my samples. These samples are from invivo animal infection experiment and some samples had to be resequenced to get enough reads. I do not have a case/infected and control/uninfected scenario in this experiment, they are just all infected samples. The range of library size in my experiment is 100,000 to 81 million. I generated a PCA plot and colored them by the sequencing depth and wanted to check with the experts if this seems to be a case wherein samples with similar sequencing depth are clustering together? Also, I tried filtering genes that have 0 reads for >50% of the samples and replotted PCA, but it looked exactly the same.
Greatly appreciate help in this regard.
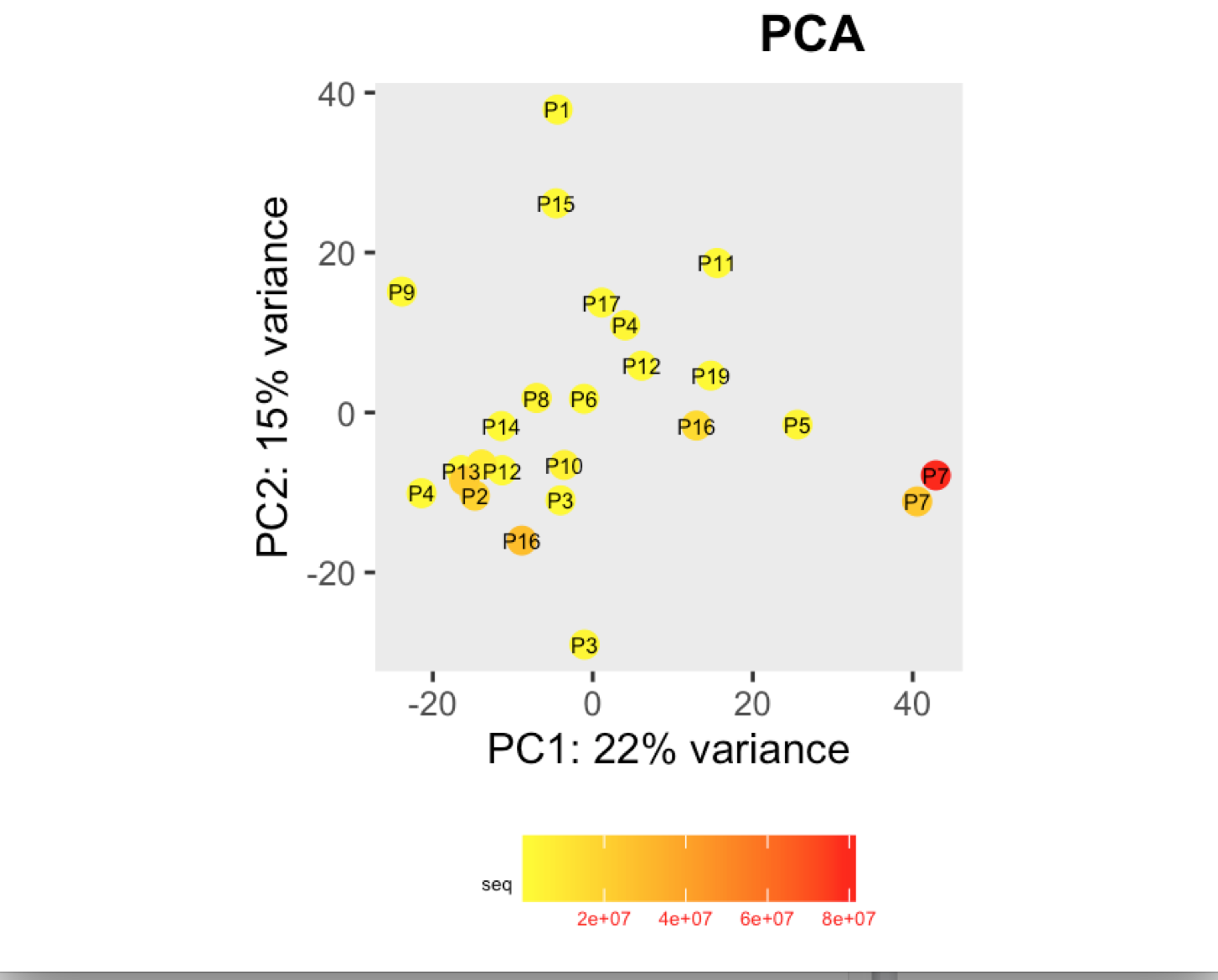

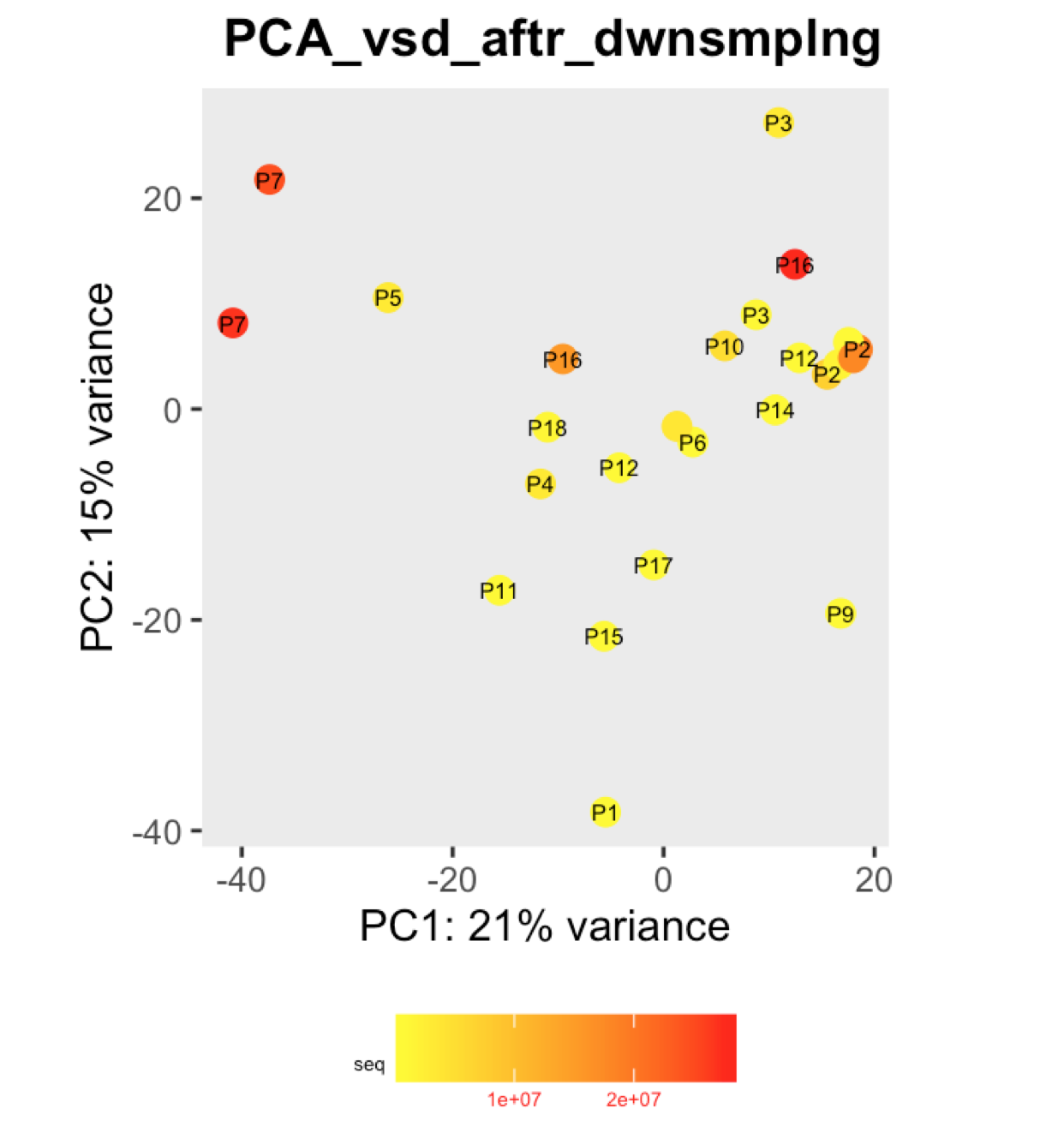
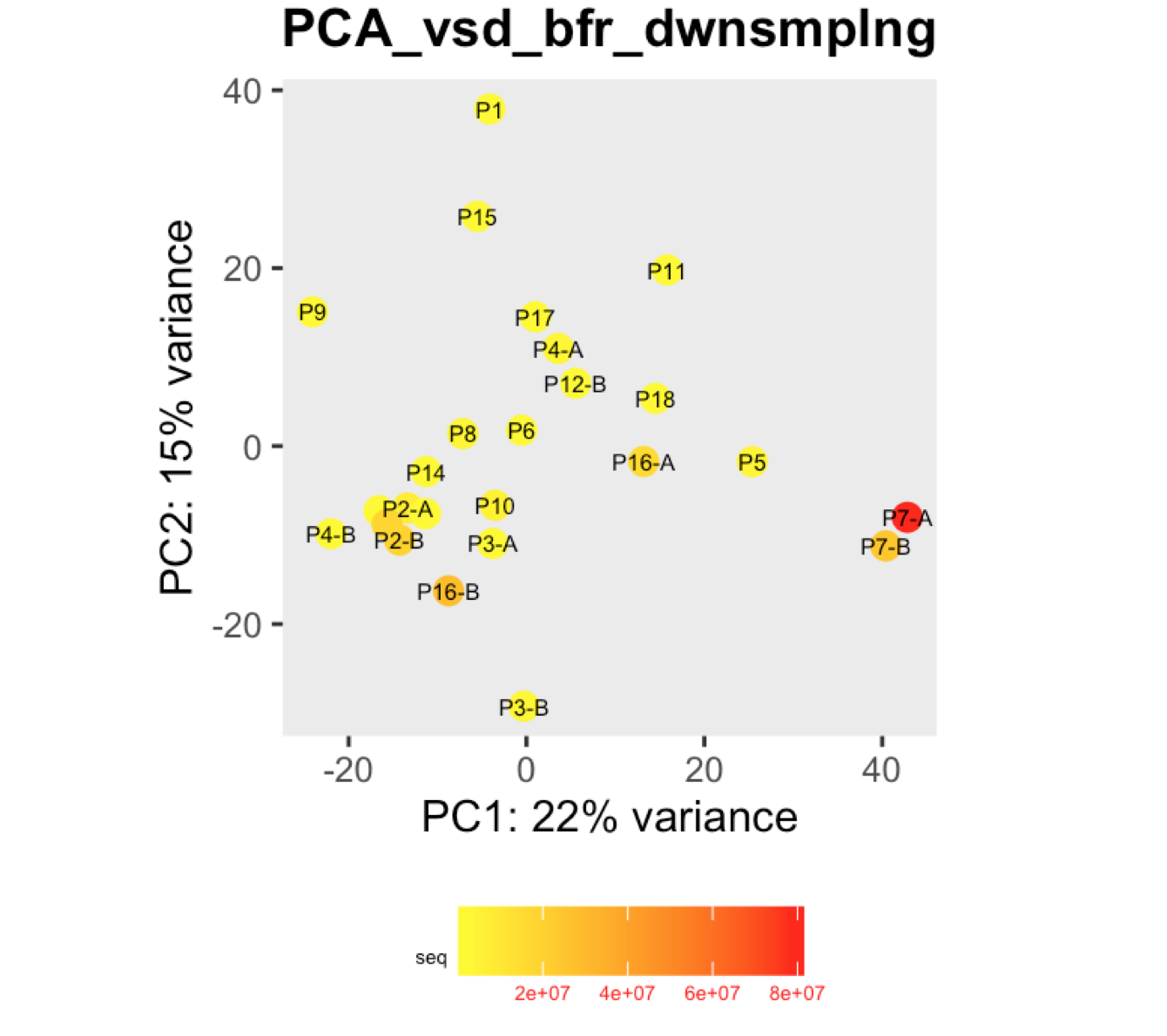
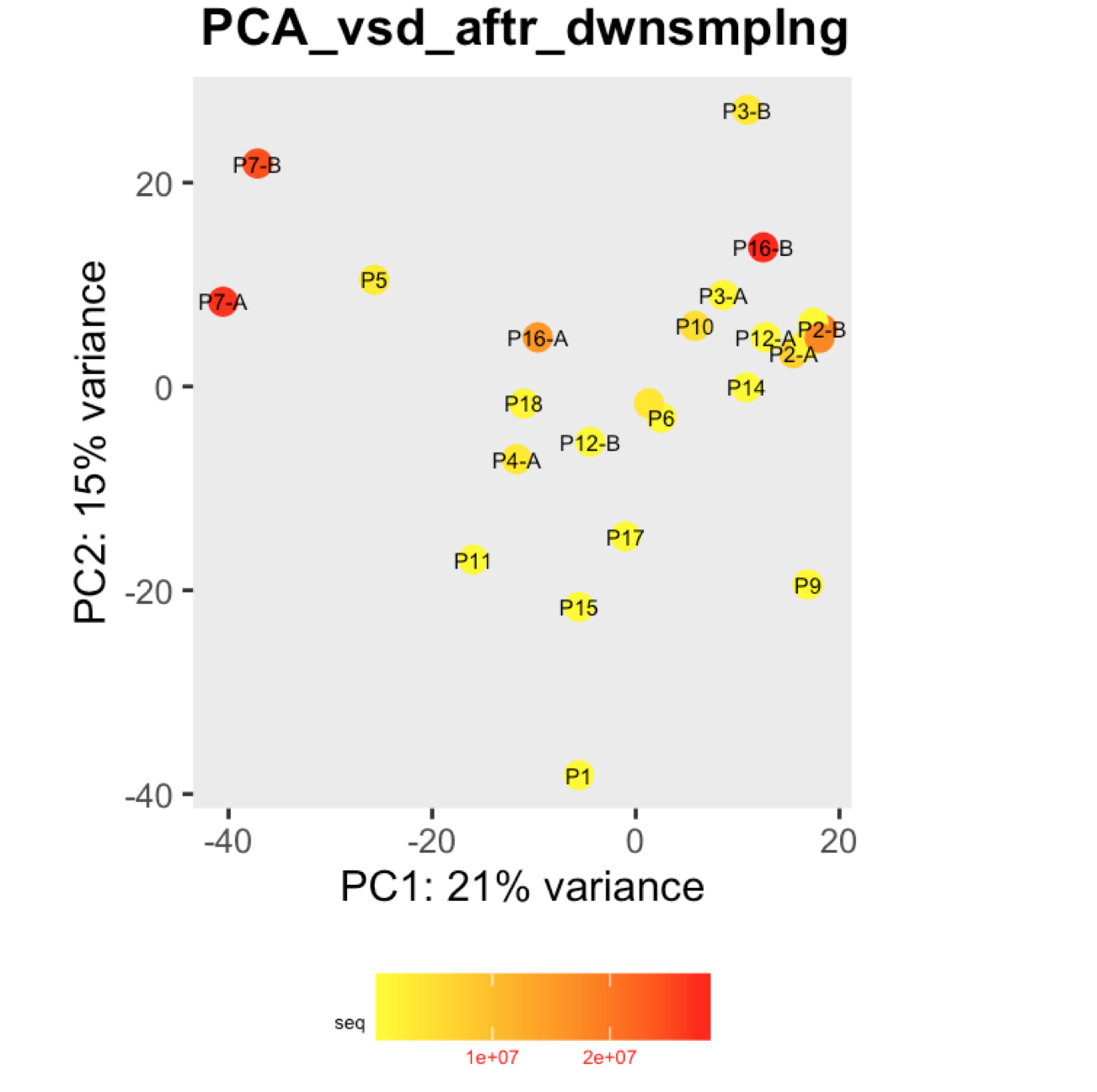
To me it doesn't look like it. Couple of notes though: (i) if you end up doing any sort of differential expression analysis, you might want to add the sequencing batch of the libraries as a control (and colour those in a separate PCA) to exclude batch effects, if any exists; (ii) the range of library sizes is quite large. You might need to take special care when analysing.
Thank you Antonio for your suggestions. Greatly appreciate it.