
We have performed RNAseq on a lot of samples of individual flies (~ 20 per condition). After running DESeq and then checking various contrasts (with ihw=TRUE), I find myself with quite a bit of genes that are detected as differentially expressed between 2 conditions. But when I plot their counts, I notice that for a lot of them, it comes from the fact that most individuals have 0 counts while 1 or 2 have a high counts, which ends up being responsible for the significance of the Wald test.
I have read that pre-filtering was recommended, something along the lines of
keep <- rowSums(counts(dds) >= x) >= y
However if you look at the example I am joining to this post 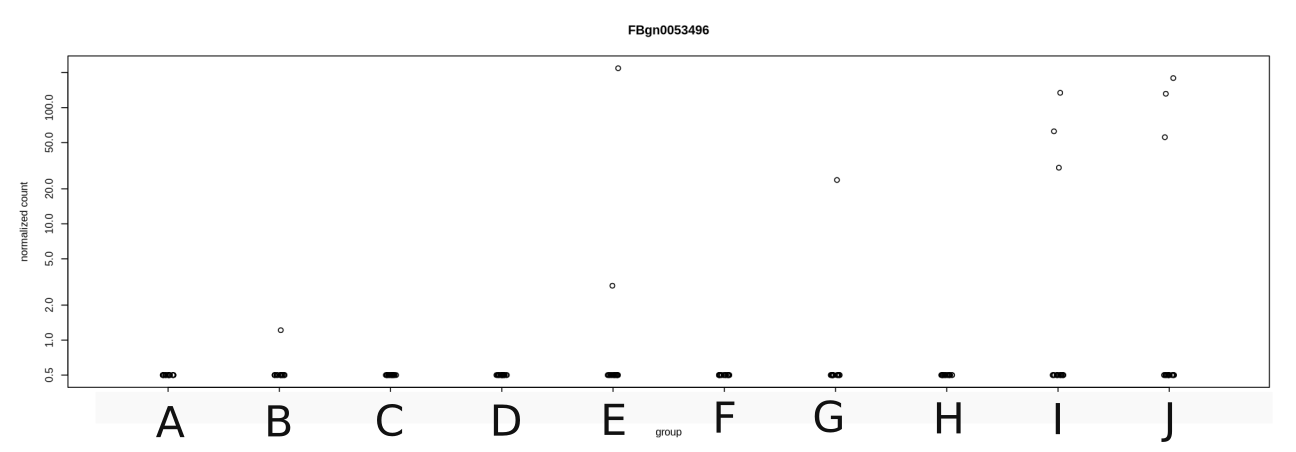
What could I do ?
Thank you !