Hi,
I am conducted a Differential expression analysis using DESeq2 to investigate the impact of treatment across sexes. The dataset includes two treatment groups, A and B. To assess sex-specific treatment effects, I am using these two approaches: (1) using the full dataset with a model that included sex and treatment as factors, followed by extraction of sex-specific contrasts; and (2) subsetting the dataset by sex and performing DESeq2 analysis separately for each group before extracting treatment-specific results. My PCA analysis does not show any separation by sex.
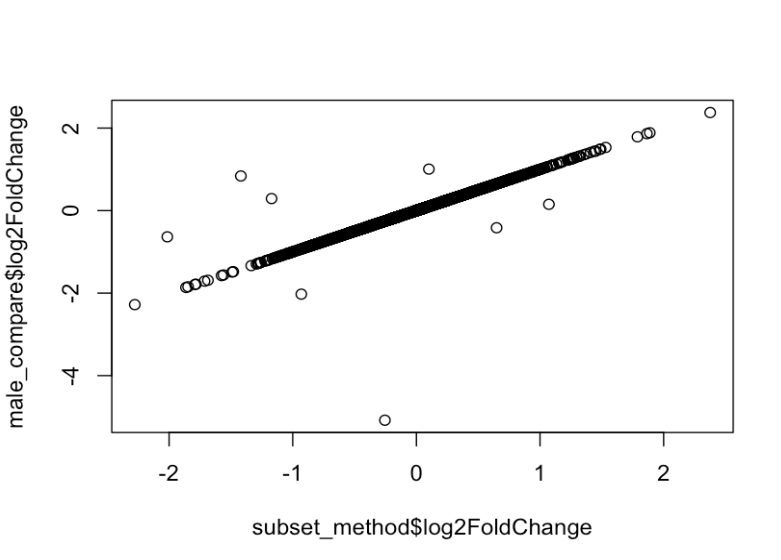
When comparing the log2 fold changes obtained from both approaches, the results were largely consistent, with most genes showing similar values. However, a few genes exhibited notable differences. Interestingly, one of these genes was the top hit in the DE analysis using the full dataset, yet it was not even statistically significant when using the subset-based approach. This discrepancy is not entirely unexpected, as p-values can vary between approaches due to differences in sample size and model structure. I also see for a few genes the baseMean is drastically different between both approaches.
I would like to understand what approach should be taken in such cases.
Whole Data
pheno <- pheno %>%
dplyr::mutate(sex_specific = paste(cohort,updated_sex, sep='_')) %>%
dplyr::mutate(sex_specific = as.factor(sex_specific))
dds_sex <- DESeqDataSetFromMatrix(countData = round(filtered_data),
colData = pheno,
design = ~0 + sex_specific)
dds_sex <- DESeq(dds_sex, parallel = TRUE)
male_compare <- results(dds_sex, contrast = c('sex_specific','treated_A_M','treated_B_M')) %>%
data.frame() %>%
dplyr::filter(!is.na(padj)) %>%
dplyr::arrange(padj) %>%
rownames_to_column(var='ensembl')
Subset Data
dds <- DESeqDataSetFromMatrix(countData = round(filtered_data),
colData = pheno,
design = ~0 + updated_sex + cohort)
dds <- DESeq(dds, parallel = TRUE)
dds_men_sub <- subset(dds, select=colData(dds)$updated_sex == 'M')
design(dds_men_sub) <- formula(~0 + cohort)
dds_men_sub <- DESeq(dds_men_sub, parallel = TRUE)
subset_method <- results(dds_men_sub, contrast = c('cohort','treated_A','treated_B')) %>%
data.frame() %>%
dplyr::filter(!is.na(padj)) %>%
dplyr::arrange(padj)
subset_method <- subset_method[male_compare$ensembl,]
plot(subset_method$log2FoldChange, male_compare$log2FoldChange)