
Hello,
I was curious if anyone has any has any tips to identifying a composition vs. efficiency vs. trended bias in their data.
To preface this (because the users guide does cover this, and is helpful), our lab ran a ChIP-seq for H2AZ in three separate sequencing and IP batches. This is clearly evident on PCA as the samples cluster along dim1 primarily based on their batch identity rather than their genotype.
From our experience, we would expect a composition bias, and profile plots around the TSS and centered peaks corroborate this where we see a genotype-dependent accumulation of this histone variant, and so I began by normalizing for a composition bias. I tried a range of bin sizes (2k - 15k) as recommended by the users guide, and settled on 10k where the normalization factors weren't changing by much as I increased from there. I've attached the MA plots below, with norm.factors as follows
normFactors(Bins_Females_SampleFiltered_10k , se.out = F)$norm.factors
[1] 0.8136641 1.0391248 1.0248323 1.0111012 0.9129898 1.0366513 1.0851679 1.1113328
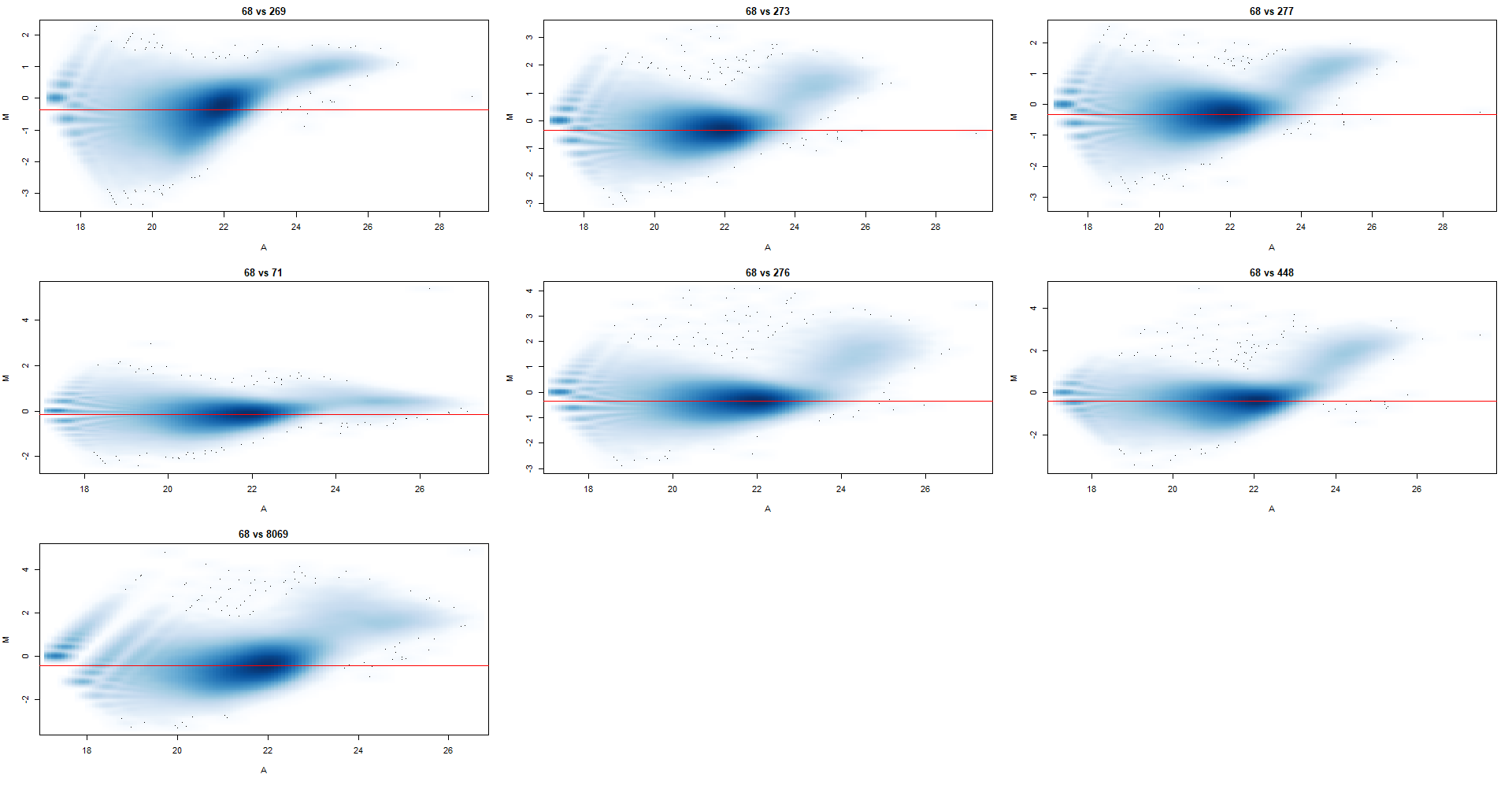
From this dataset, I've identified 2 outliers already through PCA and QC scores that have been removed (not in these MA plots). Based on the pattern of the clouds and the loess regression < 0 I would assume that there is in fact a composition bias, but what I find strange is that it's not really visible between the genotypes. i.e.: the first 3 plots are part of genotype A and the last 4 are part of genotype B, with another caveat being that sample 68 (genotype A) and 71 (genotype B) are part of one independent batch - could this be the reason that they all look different.
Additionally, after filtering windows and plotting abundance vs. FC plots, there appears to be a trended bias as well - though as far as I am aware this should be controlled for in the glmQLFit() portion of edgeR.
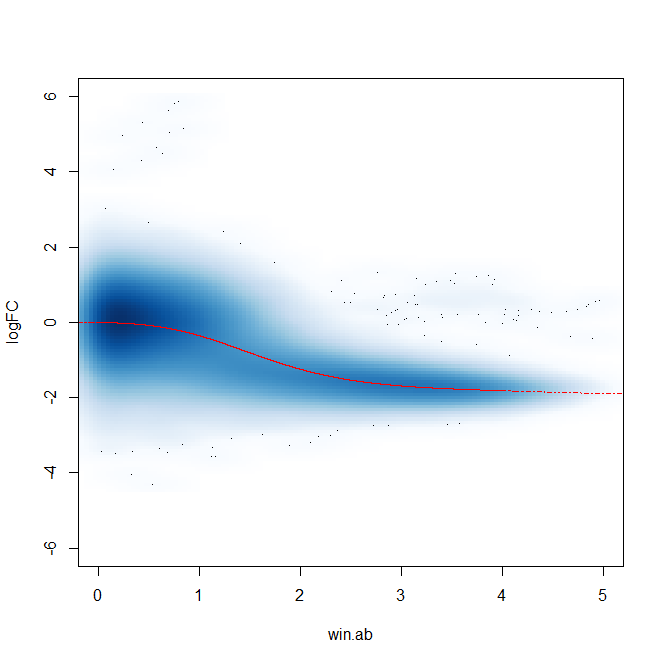
I was wondering if anyone has had issues with different sequencing batches similar to this and what they did to resolve them. I am getting the expected trend and number of DB sites and it lines up with our expectations of what's going on with this variant.

Hello Aaron,
About the IP efficiencies, yes definitely and for us, to complicate this experiment further, these batches were generated by different experimenters with three total. I have blocked for these batches in the model and batch correction with ComBat for visualization clearly demonstrate, that once the batches are accounted for, the clustering on PCA occurs in the expected genotype dependent manner along dim1 and dim2.
Regarding the difference between composition and trended bias normalization, based on our preliminary analysis, and molecular characterization (i.e.: ChIP-qPCR, Westerns, etc.) we definitely do expect a difference in accumulation, really just wanted confirmation that my logic was correct in using the composition bias given the confounding variables with these batches - we've never sequenced/IP'd something three separate times for one experiment.
Thank you very much for your help, and I really enjoy using Csaw its a fantastic tool!
Best, Luca
Thank you so much, you saved my day.