
Hi there,
I have a total of 6 donors, 2 cell types and 3 conditions (ex vivo, unstim, and stim). I run the experiments in 2 days. The donors in day one are not the same donors in day 2. In each day, I did have all 3 conditions and both cell types, so it is balanced.
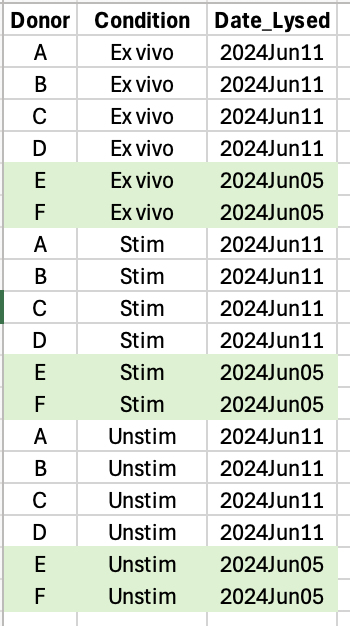
When I vsd <- vst(dds, blind=TRUE) and plotPCA(vsd, returnData = TRUE, intgroup = c("Date_lysed", "Conditions")) after running DESeq2::DESeqDataSetFromHTSeqCount(design = ~ Subset * Conditions), Stim and Unstim cells for both cell types were grouped away from ex vivo, but within unstim and stim group, the days of running experiment was the main force separating them into 2 groups.
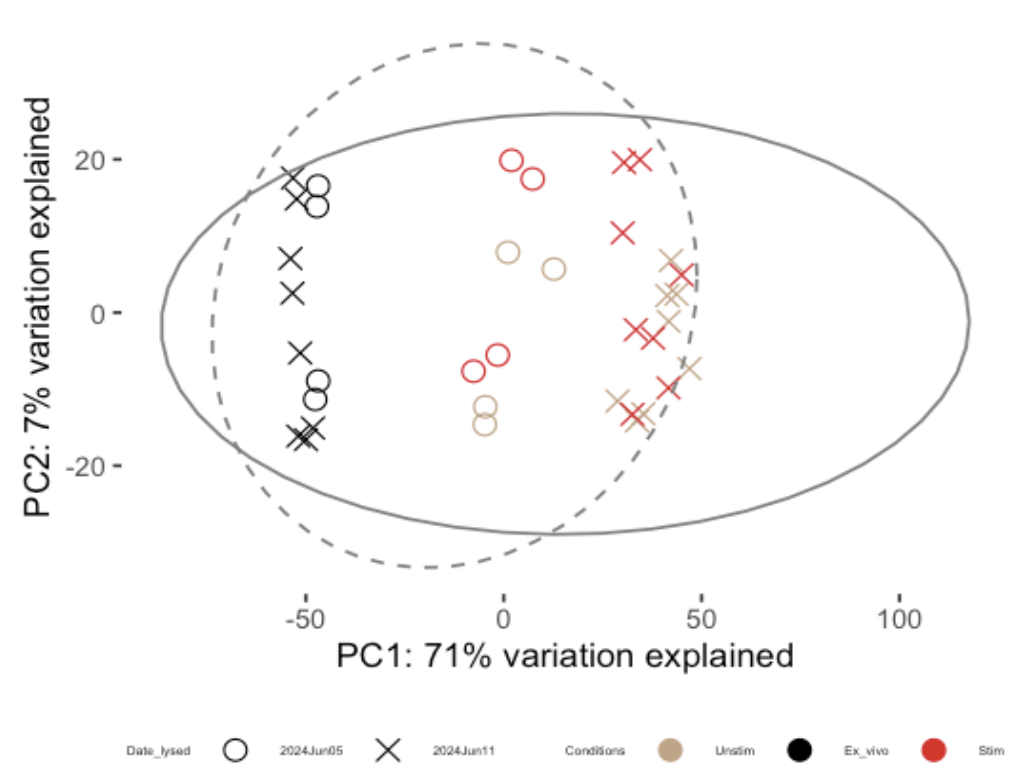
I then ddsHTSeq <- DESeq2::DESeqDataSetFromHTSeqCount(design = ~ Date_lysed + Subset * Conditions), and reran thevst(blind=FALSE)andplotPCA` on the new object, but the plot is exactly the same.
I plan to try SVA and RUV next, but anyone has any suggestions/insights?
Thanks for your help!


Hi, James. I forgot to mention, but yes, I did run
DESeq2::DESeq(). So, I ranDESeq2::DESeqDataSetFromHTSeqCount, thenDESeq2::DESeq(), thenvst(dds, blind=FALSE), thenplotPCA.From the PCA plots, I thought there was actually batch effect by
Date_lysedwithin the coloured dots (brown are unstim and red are stim)? It is because these brown and red symbols are grouped into 2 groups that I showed as 2 boxes below (circle = day 1 of experiment and cross = day 2 of experiment). Or, am I missing something about this PCA plot?Subset variable refers to two cell types for each condition. For example, ex vivo for a donor was sorted into Subset 1 and Subset 2. Subsets are not visualised in this plot. Donor is biological replicates. There is no technical replicate.
My aim is to compare differences between these 2 subsets within Ex vivo condition. In addition, I want to compare Subset 1 between Unstim and Stim, and Subset 2 between Unstim and Stim. That was why the
design = ~ Subset * ConditionsGiven the ex vivo (black crosses and black dots) do not seem to be affected by batch variable date_lysed, do you think it is best to just use the count file data of ex vivo samples to compare between Subset in ex vivo condition, then do a separate analysis with the stim and unstim samples?
Thank you so much!
How you analyze your data is up to you. But do note that Subset * Conditions implies that you want to find genes that respond to stimulus differently, depending on the cell type, which is different from what you describe. If you do care about interactions, it is IMO much easier to reparameterize to condition_subset and fit a cell means model, then make whatever comparisons you want. There is a small paragraph in the
DESeq2vignette that talks about this, but a clearer explanation IMO is in the limma User's Guide in 9.5.2-9.5.3, starting on page 45.Also, you probably want to include a Donor factor in your model to account for Donor-specific differences. But if you do so, you will not be able to include Date_Lysed, because that's nested in Donor.