
Greetings to those familiar with Feature Counts,
We have one RNA sequencing, paired-end, 150bp file for which we estimate gene and junction counts using Feature Counts. We run Feature Counts in Linux.
Feature Counts version: 2.0.8 Feature Counts code:
$FCOUNTS -a GCF_000001405.40_GRCh38.p14_genomic.gtf -G GCF_000001405.40_GRCh38.p14_genomic.fna -T 8 -J -M -s 0 -p --countReadPairs --largestOverlap -o outfile infile.bam
We have documented the following instance in a junction counts output file (outfile.jcounts) :
Genes:
PNKD TMBIM1
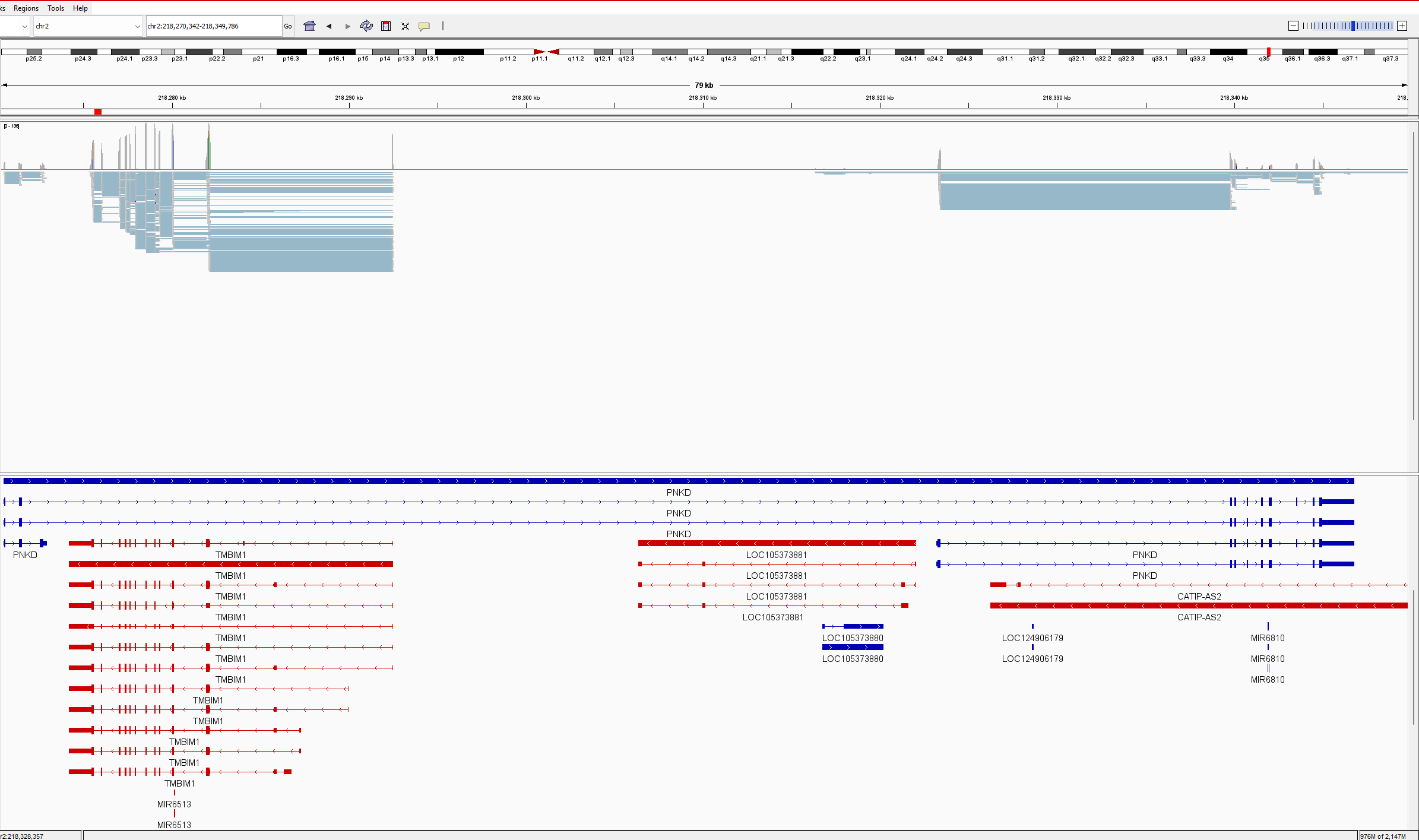
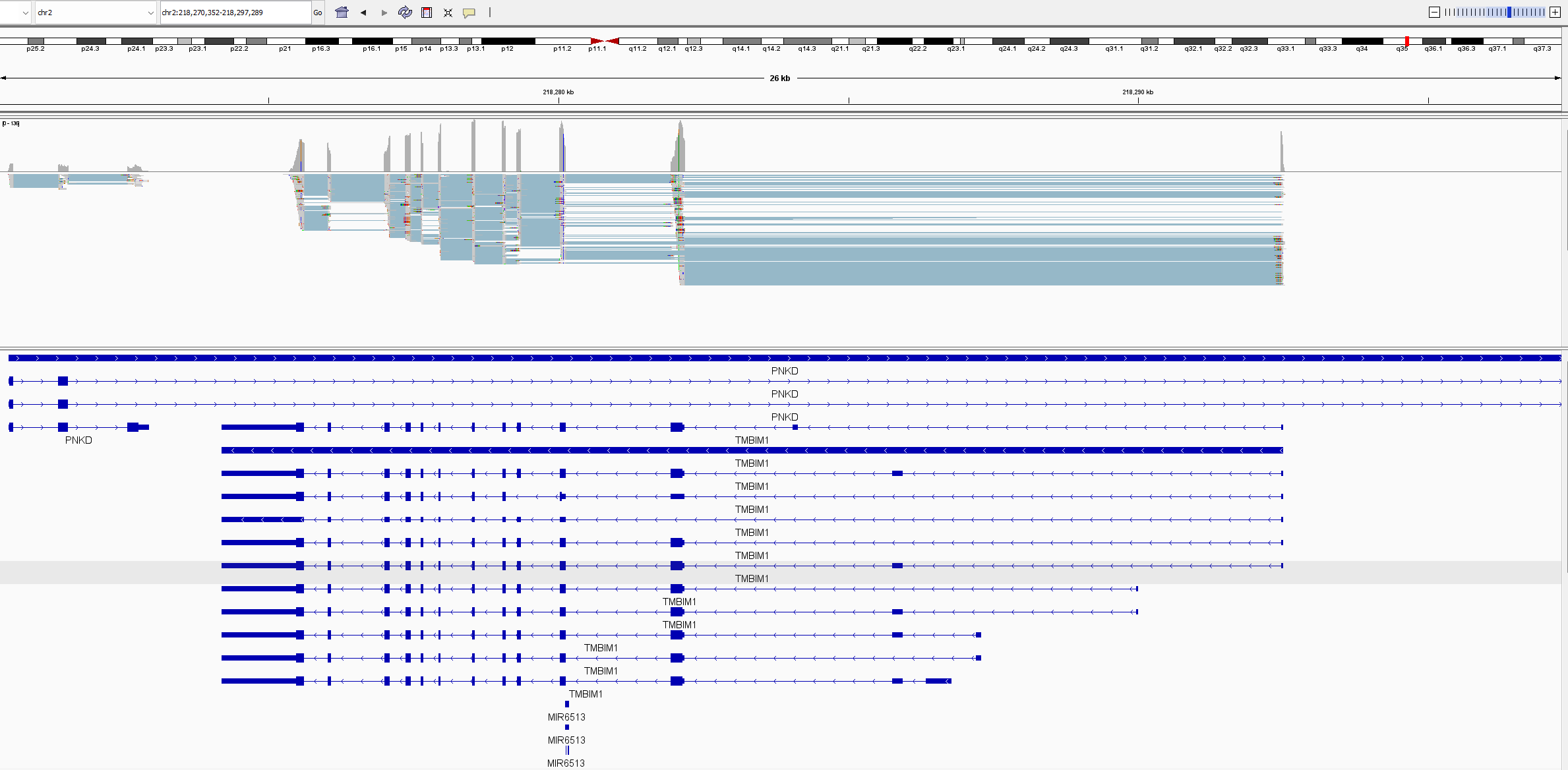
Situation (see attached picture in IGV):
PNKD gene overlaps TMBIM1 gene, and all transcripts. All TMBIM1 transcripts are in the PNKD intron. All TMBIM1 junction reads ~1000 are splitting between PNKD and TMBIM1 for multiple intron junctions. In outfile.jcounts PNKD is the primary gene, however has no exons located in this region. When we remove the PNKD gene from the GTF and recount with Feature Counts, the reads are assigned perfectly to TMBIM1, as the primary gene with NA as secondary gene, as it should. A check of gene counts using exon as feature correctly assigns counts to genes. This appears to be only a junction counting issue. The .gtf and .fna are Refseq 1405.40 files (as noted above) and are untouched. We use this as a simple verification of raw junction counts before analysing PSI. As you will also note, the reverse is also true with PNKD exon-exon junctions sharing read counts with a partially overlapped gene CATIP-AS2. We have encountered others and believe this may be a widespread event.
Here is the outfile.jcounts of that region:
PNKD NA NC_000002.12 218270602 + NC_000002.12 218271381 + 17
PNKD NA NC_000002.12 218270602 + NC_000002.12 218271424 + 4
PNKD NA NC_000002.12 218271549 + NC_000002.12 218272570 + 14
PNKD TMBIM1 NC_000002.12 218275621 - NC_000002.12 218276026 - 74
PNKD TMBIM1 NC_000002.12 218276079 - NC_000002.12 218277004 - 46
PNKD TMBIM1 NC_000002.12 218277099 - NC_000002.12 218277366 - 90
PNKD TMBIM1 NC_000002.12 218277453 - NC_000002.12 218277633 - 96
PNKD TMBIM1 NC_000002.12 218277670 - NC_000002.12 218277935 - 73
PNKD TMBIM1 NC_000002.12 218277974 - NC_000002.12 218278515 - 123
PNKD TMBIM1 NC_000002.12 218277992 - NC_000002.12 218278515 - 1
PNKD TMBIM1 NC_000002.12 218278118 - NC_000002.12 218278515 - 2
PNKD TMBIM1 NC_000002.12 218278565 - NC_000002.12 218279038 - 128
PNKD TMBIM1 NC_000002.12 218279091 - NC_000002.12 218279289 - 101
PNKD TMBIM1 NC_000002.12 218279091 - NC_000002.12 218280026 - 6
PNKD TMBIM1 NC_000002.12 218279353 - NC_000002.12 218280026 - 109
PNKD TMBIM1,MIR6513 NC_000002.12 218280126 - NC_000002.12 218281940 - 26
PNKD TMBIM1,MIR6513 NC_000002.12 218280126 NA NC_000002.12 218292466 NA 34
PNKD TMBIM1 NC_000002.12 218282181 - NC_000002.12 218284039 - 1
PNKD TMBIM1 NC_000002.12 218282181 - NC_000002.12 218285763 - 1
PNKD TMBIM1 NC_000002.12 218282181 - NC_000002.12 218287207 - 1
PNKD TMBIM1 NC_000002.12 218282181 NA NC_000002.12 218292466 NA 71
PNKD LOC105373881 NC_000002.12 218316364 + NC_000002.12 218385147 + 3
PNKD LOC105373880,LOC105373881 NC_000002.12 218316883 + NC_000002.12 218317955 + 2
PNKD CATIP-AS2 NC_000002.12 218323428 NA NC_000002.12 218341591 NA 5
PNKD CATIP-AS2 NC_000002.12 218323431 + NC_000002.12 218339783 + 52
PNKD CATIP-AS2 NC_000002.12 218339898 + NC_000002.12 218340029 + 27
PNKD CATIP-AS2 NC_000002.12 218340141 + NC_000002.12 218340728 + 8
PNKD CATIP-AS2 NC_000002.12 218340786 + NC_000002.12 218341534 + 5
PNKD CATIP-AS2 NC_000002.12 218341626 + NC_000002.12 218341981 + 12
PNKD CATIP-AS2 NC_000002.12 218342144 + NC_000002.12 218343500 + 12
PNKD CATIP-AS2 NC_000002.12 218343586 + NC_000002.12 218344455 + 15
PNKD CATIP-AS2 NC_000002.12 218344570 + NC_000002.12 218344808 + 28
Has anyone encountered this before and know where things have gone wrong? Prof Smyth suggested we post here instead of Biostars, to hear some constructive feedback from the Subread authors.
Thanks in advance, Chris.


When you use
featureCountsto count junctions with the-Joption, it doesn't check if the junction is exactly overlap with exons of genes. Instead, it looks at the whole gene regions (from the first base to the last base of each gene). If the junction is anywhere within the region of a gene, it's assigned to that gene.Hi Yang Liao, thanks for the note.
We use Feature Counts extensively for gene-level counting (exon features).
Unfortunately though, and I'll wait for other people's professional opinion to comment on this further, from your comments I disagree with the definition of the function and how it works.
I feel users should not have to go and check whether the splice junctions indeed exist in every primary gene of the list (now that I know it is not tracking junction counts as per the annotation).
Further, there are possibly 15 000 occurrences of this, in this one bam file aligned to Refseq 1405.40 . Gencode has similar overlapped genes on the same strand and opposite strand.
Also in the example provided, the junction data rows correctly annotate the strand and coordinates of TMBIM1 (strand negative) in site 1 and site 2, not PNKD (strand positive). The only column recording any information relating to PNKD is the primary gene column.
Other programs annotate and count the splices correctly using 2 exons and corresponding transcript IDs, and the transcript IDs' corresponding Gene Id's to build the junction correctly. (Never using the gene start and end coordinate to act as a bucket for any junctions located in the region).
While it was extremely convenient and useful to use -J and obtain junction counts at the same time as our gene counts, now that I know it is incorrect and I will no longer be able to this feature. We almost submitted this data to external reviewers and it would have been incorrect. However, we counted the junctions with several programs and documented the discrepancy.
Having said of all the above, we hope the moderators agree there is an error here that should be addressed because having this feature in FeatureCounts is extremely useful.
Thanks for considering, Chris.