
Hi everyone,
I'm currently analyzing ~30 treatments vs mock using DESeq2 (v.1.44.0). I don't filter any low counts. I apply L2FC-shrinkage (ashr).
This is an experiment where we repeated the library prep from exactly the same RNA which was used for a first prep/experiment some weeks ago.
I will use synonym for treatments and genes but I think it's clear what I mean. Here, I extracted the results for a given gene A in treatment1 vs. mock frm two independent experiments.
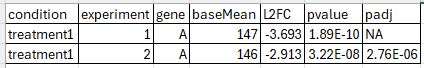
Why is gene A not a DEG after multiple testing correction in experiment 1, but in experiment 2 although it has comparable baseMean and even stronger L2FC and lower p Value?
What are other potential features that can influence the adjustment of the p-val adjustment in such a way that I'm ending up with a DEG in one experiment but not in the other? Number of replicates is identical, total number of treatments in the experiment is identical.
Thanks!
It would probably help if you broke out the raw and normalized read counts for all the samples.
As said, literally identical on a continuous scale but different in categorical terms.
I think you run into the common pitfall that you treat results as categorical (significant, not significant) whereas in reality the results are almost identical. Look at the baseMean, logFC and pvalue, they're close. The independent filtering removed this gene (reason unknown, maybe because counts / baseMean is relatively low. This makes the difference here. If you feel this is inappropriate you could run the analysis with some low count filtering (see vignette) and then turn off the independent filtering.
I would say baseMean, read counts, L2FC, ... are very similar between the two experiments - that's why I was wondering about the results.
With "reason unknown" you mean it's not easily possible to trace back why exactly gene A is DEG in one experiment, but not in the other? So in the end it's a mix of baseMean, L2FC, replicates per treatment, deviation across replicates, number of samples in the experiment, etc?
I was also reading on the independent filtering and will give it a try. Thanks.
Unknown reason means that from the results you show one cannot easily infer why exactly the IF removed this gene in one but not in the other condition. If you turn IF off you will probably be fine, but still, hard cutoffs can induce differences even though data are very similar, that is known.
You could check the reply I got to a similar issue here: Adjusted p-values become NA when sub-setting samples
In short, DESeq2 uses by default
independentFiltering=TRUE. This creates a separate baseMean threshold for each pairwise comparison you run. Then, if you want to compare the results/DEG between multiple pairwise comparisons, some of them might have much higher baseMean thresholds than others. This leads to genes suddenly getting padj=NA.AFAIK, the advised way is to disable
independentFilteringand apply the same (manual) baseMean threshold to all comparisons.