Entering edit mode

Hi folks,
I am trying to do a KEGG pathways using pathview in R.
Example of data
data<- data.frame(
EntrezID = c(107965807, 406114, 410747, 409341, 727522, 113218875, 410228, 113219265, 100576126, 102656882),
d3 = c(-1.8505053, -1.6865244, -1.5905482, -1.5222310, -1.4876187, -1.4257670, -1.3510711, -1.3431686, -1.3236289, -1.3113374),
d1 = c(NA, -1.0918122, -1.1389847, -0.9339650, NA, -1.1790280, -1.1046071, -1.2304113, NA, -0.9977286)
)
Example with one state (using gene list)
d3 - extracting logFC and names for d3
geneList1=data$d3 # logFC (negative and positive)
names(geneList1)=data$EntrezID # entrezid for each gene
Run Pathview
pathview(gene.data = geneList1,
pathway.id = "ame04145",
species = "ame",
na.col = "transparent",
low = list(gene = "blue", cpd = "blue"),
mid =list(gene = "pink", cpd = "pink"),
high = list(gene = "red", cpd ="red"),
limit = list(gene = 3),
bins = list(gene = 10),
both.dirs = list(gene = T),
out.suffix="_d1"
)
Very nice plot with gene modulated colored as asked
Comparing two states (d1 and d3)
rownames(data)=data$EntrezID
pathview(gene.data = data[,2:3],
limit = list(gene = 3),
bins = list(gene = 10),
both.dirs = list(gene = T),
pathway.id = "ame04145",
species = "ame",
keys.align = "y",
kegg.native = T,
match.data = T,
same.layer = T,
multi.state = T, out.suffix="mixed")
Output
Warning: None of the genes or compounds mapped to the pathway!
Argument gene.idtype or cpd.idtype may be wrong.
Warning: No annotation package for the species ame, gene symbols not mapped!
Info: Working in directory /Users/camilledeboissel/Library/CloudStorage/OneDrive-Personal/Post-doc/Amro-Zayed_BeeCSI/Phiphi_analysis/All_samples_analysis
Info: Writing image file ame04145.mixed.png
Warning message:
In colnames(plot.data)[c(1, 3, 9:ncs)] <- c("kegg.names", "all.mapped", :
number of items to replace is not a multiple of replacement length
What am I doing wrong here? Why is it working with a geneList input and not as multistate ?
Thank you for your help
A quick comment after I copied/pasted your code: when using
geneList1in the 3rd code chunk I do not get a nice plot... I rather got the same error that none of the ids mapped to the pathway! Yet, it does work when mapping topathway.id = "ame00500"! Note that in your code chunk you used as inputgeneList(thus without the suffix 1!)Thus: your example input can indeed not be mapped on pathway
ame04145.Thank you! Here a subset of genes that really works with "ame04145".
I run
And this i my actual output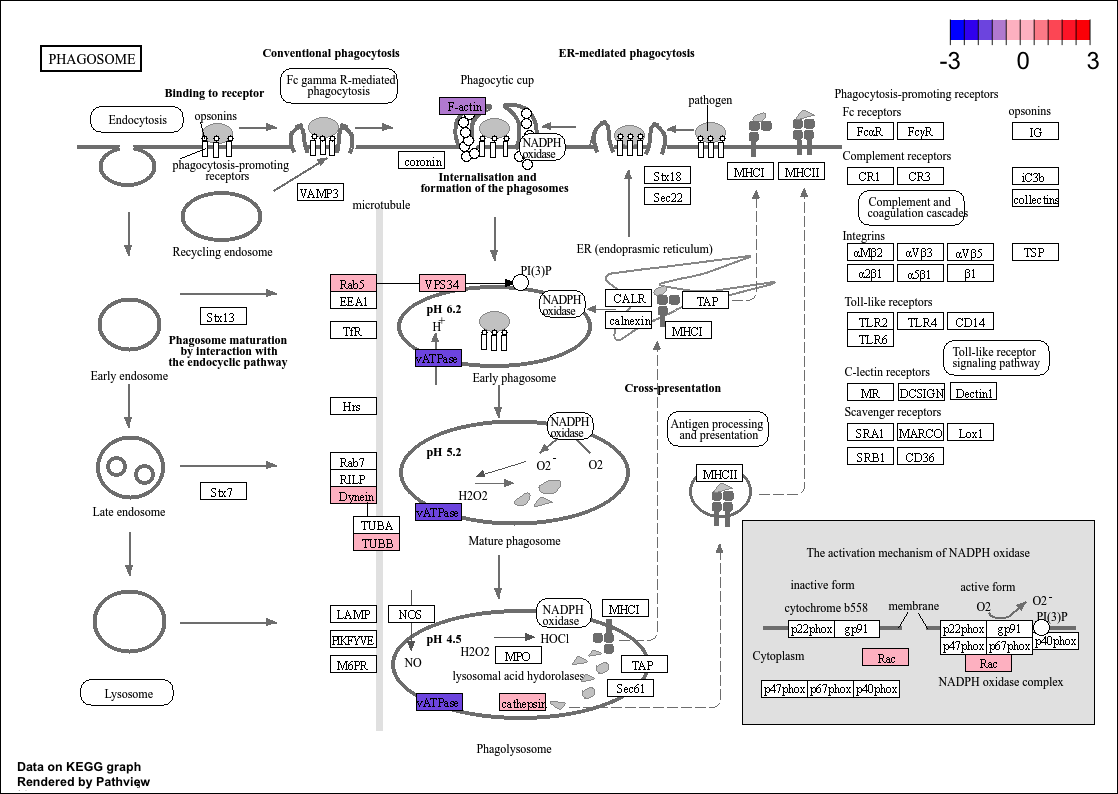
When I run
And I got this pawthay...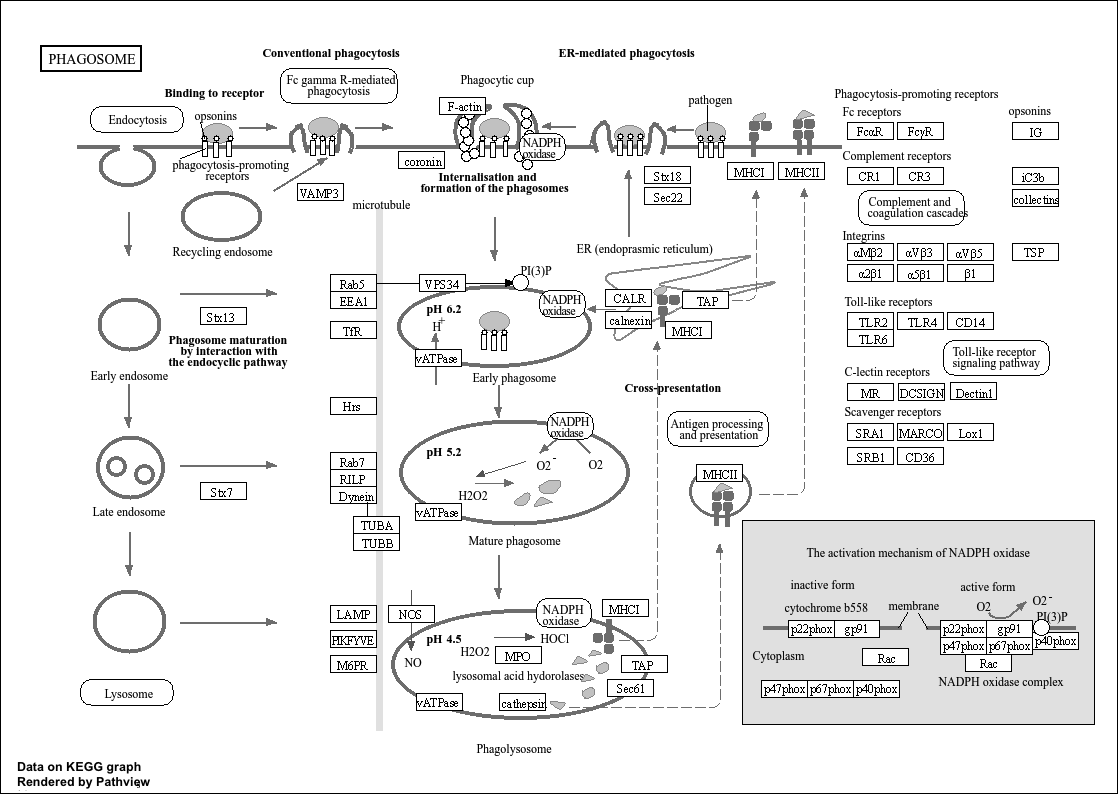
So i am not sure what I am missing here. Thanks for your feedback
According to the the help page of
pathview(type?pathview) the input should beeither vector (single sample) or a matrix-like data (multiple sample). Note that your input is adata.frame. Converting this to amatrixmakes your code work, taking into account theNA.:D Thank you!!! It works perfectly.