
I am counting reads from rna-seq with htseq-count using a gtf file of enhancer regions, therefore, i want to see how much rna is transcribed from the enhancer.
My problem is that the amount of counts is very low and I obtain a count table like this:
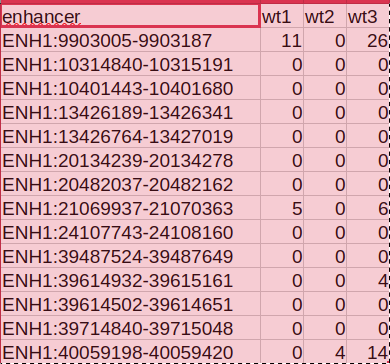
My supervisor is telling me to do a binary table like this, where i have to do the mean (?) of my counts and then establish different column count thresholds and just check if my enhancer is over that value (1) or not (0). Then i have to compare this data with a known enhancer clasification vector with 0 and 1 and see the best coincidence between my rna data and this vector
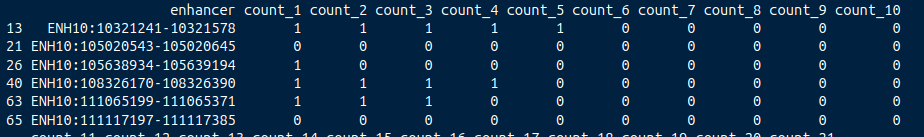
But always my best count threshold is on the extreme values (count_0 or count_max) so i guess i am not doing something correctly
What could i do?
Hi, your question is off-topic here since it does not directly concern a particular Bioconductor package. Maybe posting at biostars.org will give you a more general audiance. In any case, I would strongly advise against binary analysis based on hard cutoffs. It inflates differences when values are just one count below or above threshold, but in fact data are actually almost exactly the same. Also, be careful comparing regions across the genome. Amplification-, GC- etc biases are very different between loci, making within-sample comparison hard. If you post at biostars, be sure to write down in more detail what exactly your question is. It is by the way known that enhancer RNA levels are much lower compared to mRNA, so low counts are expected.