
Dear Bioconductor community,
I have a question about limma
First, let me explain about my data.
T0 (the starting point of medication) and T4 (the time point when DNA methylation data was obtained from blood samples) are separated by a span of two years.
We coded individuals who took lithium for two years as 'cases' and those who did not as 'controls,' assigning each group an Age_group value of 1 or 0, respectively.
[data] result (data.frame)
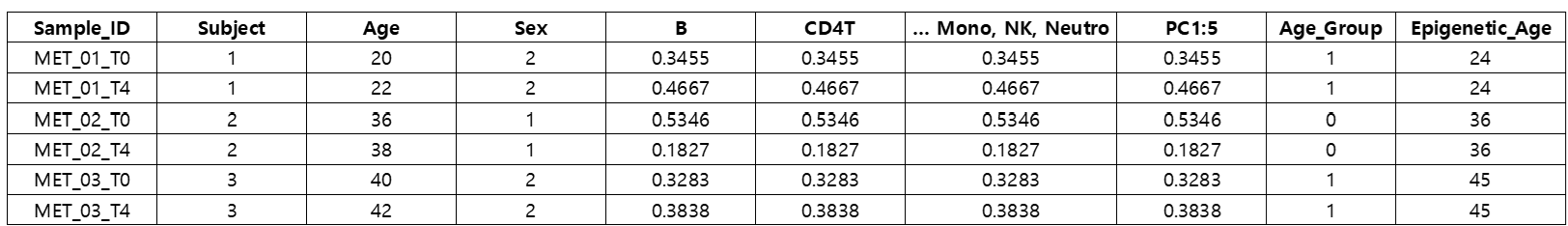
The goal of this analysis is to examine whether lithium treatment could reduce the Epigenetic Age predicted from DNA methylation data.
The covariates included in the model are Age, Sex, PC1, PC2, PC3, PC4, PC5, B, CD4T, CD8T, Mono, NK, and Neutro (cell type proportions).
Initially, I simply used
lm(Epigenetic_age ~ Age_group + Age + Sex + PC1 + PC2 + PC3 + PC4 + PC5 + B + CD4T + CD8T + Mono + NK + Neutro)
to observe the association.
However, to achieve a more accurate model, I implemented model.matrix directly based on the guidelines from the following source: https://bioconductor.org/packages/release/workflows/vignettes/RNAseq123/inst/doc/designmatrices.html#treating-factors-that-are-not-of-direct-interest-as-random-effects.
I intended to include individual as a random effect ,therefore, I grouped the samples based on Group) Age_group_Time
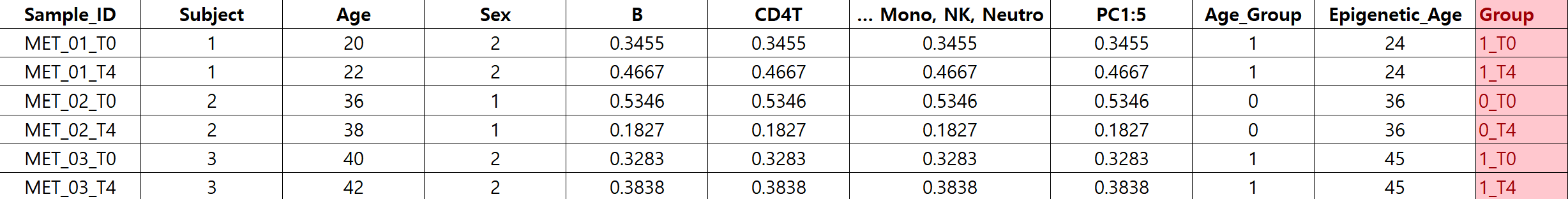
and created the model.matrix accordingly
design <- model.matrix( ~ 0 + Group + AGE + SEX + BMI + ALCOHOL_USE + SMOKER + BD_Type + batch + Acute_Stable + Center + PC1 + PC2 + PC3 + PC4 + PC5 + B + NK + CD4T + CD8T + Mono + Neutro , data = result)
And then, I ran the lmfit and set up a contrast matrix for comparison.
fit<- lmFit(object = result$Epigenetic_Age, design, block = Subject, correlation = cor$consensus.correlation)
contrasts_Eigenetic_Age <- makeContrasts(
R_T4vsT0=Group1_T4-Group1_T0,
NR_T4vsT0=Group0_T4-Group0_T0,
RvsNR=(Group1_T4-Group1_T0)-(Group0_T4-Group0_T0),
levels = colnames(design)
)
fit_PCGrimAge <- contrasts.fit(fit, contrasts_Epigenetic_Age)
fit_PCGrimAge$coefficients
Currently, I can only obtain coefficients as output so that I tried to further analysis with eBayes() to get P-value but I thought it is only for DE genes. (In my case I only have one (Epigenetic_Age))
Am I right? I'm not sure if it is right*
Any advice would be greatly appreciated.
Thank you.
Best
Yujin


Thanks to you, I learn a lot.
Many thanks,
Yujin.