
Hi there,
So, I'm running DESeq2 and have a number of different of different groups (>10) for comparisons. When I reach the prefiltering step, I filter for genes with a mean count above 1. Then, I also try to keep only the genes that have a normalised count greater than or above 10, in at least 10 samples.
countdata <- subset(countdata,apply(countdata, 1, mean) >= 1)
...
dds = estimateSizeFactors(dds)
keep = rowSums(counts(dds, normalized = T) >= 10) >= 10
dds = dds[keep,]
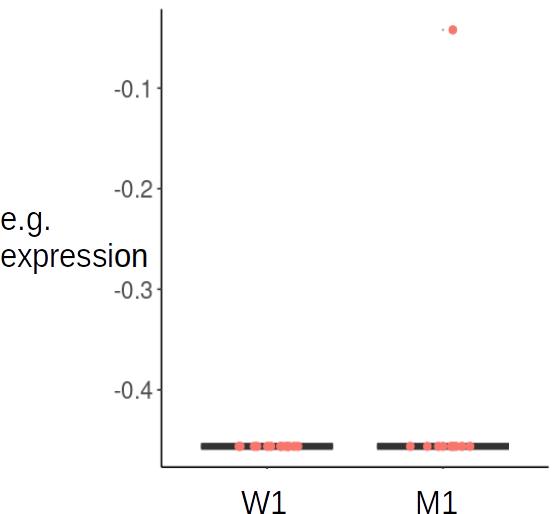
But because the filtering works for a gene in all samples at once, when I extract results for certain comparisons that exclude samples, some genes are retained that will have counts of 0 in one group (n = 12), and majority 0 in another group (n = 10) except for one or more samples that could be e.g. 3. Sort of like the example below:
ID W1 W1 W1 W1 W1 W1 W1 W1 W1 M2 M2 M2 M2 M2 M2 M2 M2 M2
Gene1 0 0 0 0 0 0 0 0 0 0 0 0 3 0 0 0 0
Due to these outliers, these genes are determined significantly differentially expressed between comparisons, and you can see it seems to be the result of these outliers.
The only similar issue I could find was this post - I cleaned the code into a working format and switched the values to filter by median rather than mean:
#Get assay data containing only the conditions of interest
assay_of_conditions =data.frame(assay(dds, "counts"))[colData(dds)$condition %in% c("Condition1", "Condition2")]
#Make a logical vector that finds the median of each row, and returns True or False based on if the median is > 3
filter_assay = apply(assay_of_conditions, 1, median, na.rm = T) > 3
#Get the results of the dds by this condition, and filter by the logical vector
res = results(dds, c("condition", "Condition1", "Condition2"))[filter_assay,]
My only question is, does this post-DESeq2 filtering seem appropriate? I worry the significance value is affected by running without pre-filtering removing the problematic outliers in the first place.
Thank you!
