
I have the following experimental design:
- four developmental
stages, effectively a nominal categorical variable - three
conditions: two treatments and one control
The biggest source of variation in the data is stage. The two treatments are conceptually close to each other and the genes responding to them are largely the same, although: one of the treatments has a stronger effect (higher fold changes) and both treatments also elicit specific responses.
What I'm mainly interested in is genes responding to both treatments and genes responding to one but not the other, at each stage.
The approach I have accepted is to unite stage and condition in a single variable group and do Wald tests for contrasts treatment1_vs_control and treatment2_vs_control for each stage separately and apply a universal FDR cutoff. It feels though that the more stage×treatment combinations a gene is discovered to be differentially expressed in, the higher the chances of eventual sub-significant tests to be true discoveries in other combinations as well, which is something I do not take into account. For example, the following gene:
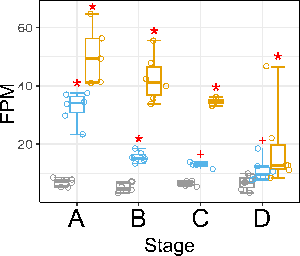
passes my FDR cutoff of 0.05 (the red asterisks) at two stages in treatment1 (blue) and at all stages in treatment2 (yellow), but the sub-significant test for stage C (raw P ~ 0.002) turns out not be significant although the totality of the evidence points to it being differentially expressed here as well albeit with a lower fold change.
My ad hoc idea of dealing with this, is to use two FDRs, say 0.1 and 0.05 in a two-step procedure: if a gene is discovered to be DE in more than some pre-defined number of stage×treatment combinations under the more relaxed cutoff, it is considered to be DE in all of them, otherwise the more stringent cutoff is used.
Is my intuition wrong, is there a better approach to this idea or to my design in general? Thanks!

Can your developmental stages be defines in terms of a continous time measure (like day 1-3-5 / hour 1-3-5?)? If so, you could also fit a linear (or non-linear) model within deseq for an overview of genes that overall change throughout your developmental curve. You won't get a granular A/B/C/D resolution, but I have a feeling in most cases you care about progression over time, so maybe this will work for you:
And for genes that show changes in one group and not the other, you could subset the treatment data as 2 df, run the same test separately with:
And then compare the significant subsets in both. Hope that helps
No, the developmental stages are too different from each other. In fact, based on prior knowledge and gene expression pattern, they show a strong clustering A+C and B+D. The gene I've shown in my post is not representative of the general pattern: most of the DE genes also show similar expression pattern uniting A+C and B+D instead.
Your second idea to split data in two for treatment1 and treatment2 and run LRT sounds very interesting. So given my full design
~ stage * treatment, the reduced design would be~ stage.