
Dear EPIC users,
I have question about EPIC v2. I analyzed my data using minfi and got beta values for all the probes that passed quality ect. Now I would like to create bigwig files so that I can look at my data in UCSC or another genome browser. However, that is where I encountered a problem.
This is the code I used:
library(IlluminaHumanMethylationEPICv2anno.20a1.hg38)
library(BSgenome.Hsapiens.UCSC.hg38)
library(rtracklayer)
data(Locations)
annotation <- getAnnotation(IlluminaHumanMethylationEPICv2anno.20a1.hg38)
annotation_df <- as.data.frame(annotation)
positions_EPIC=data.frame(row.names = row.names(annotation_df),
chr=annotation_df$chr,
start=annotation_df$pos,
end=ifelse(annotation_df$strand == "+", annotation_df$pos + 1, annotation_df$pos - 1),
strand=annotation_df$strand)
# Function to swap start and end if start is greater than end
swap_if_needed <- function(start, end) {
if (start > end) {
temp <- start
start <- end
end <- temp
}
return(c(start, end))
}
# Apply the function to each row of the data frame
for (i in 1:nrow(positions_EPIC)) {
result <- swap_if_needed(positions_EPIC$start[i], positions_EPIC$end[i])
positions_EPIC$start[i] <- result[1]
positions_EPIC$end[i] <- result[2]
}
# Print the updated data frame
print(positions_EPIC)
hg38info=seqinfo(BSgenome.Hsapiens.UCSC.hg38)
mybeta2 = na.omit(mybeta)
for (i in 1:ncol(mybeta2)){
mySample=merge(positions_EPIC,mybeta2[,i],by="row.names")
names(mySample)[6]="score"
mySample$Row.names=c()
print(mySample)
name=colnames(mybeta2)[i]
write.table(mySample[,c("chr","start","end","score")],
sep="\t",
row.names = FALSE,
col.names = FALSE,
quote = FALSE,
file=paste(name,".bedGraph",sep=""))
mySample$strand = NULL
print(mySample)
myRanges=makeGRangesFromDataFrame(mySample,
keep.extra.columns = TRUE)
seqinfo(myRanges)=hg38info
export.bw(myRanges,paste0(name,".bw"))
}
this worked till the very last line - export.bw
Error in FUN(extractROWS(unlisted_X, IRanges(X_elt_start[i], X_elt_end[i])), :
BigWig ranges cannot overlap
In addition: Warning message:
In valid.GenomicRanges.seqinfo(x, suggest.trim = TRUE) :
GRanges object contains 4 out-of-bound ranges located on sequences chr6,
chr11, chr14, and chr18. Note that ranges located on a sequence whose length
is unknown (NA) or on a circular sequence are not considered out-of-bound
(use seqlengths() and isCircular() to get the lengths and circularity flags
of the underlying sequences). You can use trim() to trim these ranges. See
?`trim,GenomicRanges-method` for more information.
Then I decided to skip coupe of list lines and just write all the bedgraph files (since that worked), and then I tried to convert them using bedgraphToBogwig finction from UCSC but then error was different - End coordinate 140288269 bigger than chr11 size of 135086622 line 168183 of file
And indeed in EPIC annotation this is the position of the prob/cpg
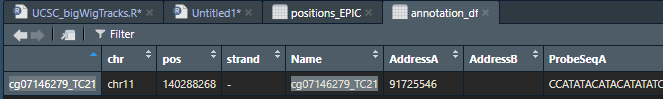
however that is out of the coordinates in hg38 genome

Does anyone knows why is this, and how to generate bigwig files?
Thank you all, Maja
> sessionInfo( )
R version 4.4.0 (2024-04-24 ucrt)
Platform: x86_64-w64-mingw32/x64
Running under: Windows 10 x64 (build 19045)
Matrix products: default
locale:
[1] LC_COLLATE=Dutch_Netherlands.utf8 LC_CTYPE=Dutch_Netherlands.utf8
[3] LC_MONETARY=Dutch_Netherlands.utf8 LC_NUMERIC=C
[5] LC_TIME=Dutch_Netherlands.utf8
time zone: Europe/Amsterdam
tzcode source: internal
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] DNAmArray_2.0.0
[2] pls_2.8-3
[3] FDb.InfiniumMethylation.hg19_2.2.0
[4] org.Hs.eg.db_3.19.1
[5] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
[6] GenomicFeatures_1.56.0
[7] AnnotationDbi_1.66.0
[8] BiocManager_1.30.23
[9] devtools_2.4.5
[10] usethis_2.2.3
[11] IlluminaHumanMethylationEPICv2manifest_1.0.0
[12] BSgenome.Hsapiens.UCSC.hg38_1.4.5
[13] BSgenome_1.72.0
[14] rtracklayer_1.64.0
[15] BiocIO_1.14.0
[16] IlluminaHumanMethylationEPICv2anno.20a1.hg38_1.0.0
[17] minfi_1.50.0
[18] bumphunter_1.46.0
[19] locfit_1.5-9.10
[20] iterators_1.0.14
[21] foreach_1.5.2
[22] Biostrings_2.72.1
[23] XVector_0.44.0
[24] SummarizedExperiment_1.34.0
[25] Biobase_2.64.0
[26] MatrixGenerics_1.16.0
[27] matrixStats_1.3.0
[28] GenomicRanges_1.56.1
[29] GenomeInfoDb_1.40.1
[30] IRanges_2.38.0
[31] S4Vectors_0.42.0
[32] BiocGenerics_0.50.0
[33] lubridate_1.9.3
[34] forcats_1.0.0
[35] stringr_1.5.1
[36] dplyr_1.1.4
[37] purrr_1.0.2
[38] tidyr_1.3.1
[39] tibble_3.2.1
[40] ggplot2_3.5.1
[41] tidyverse_2.0.0
[42] readr_2.1.5
loaded via a namespace (and not attached):
[1] splines_4.4.0 later_1.3.2 bitops_1.0-7
[4] preprocessCore_1.66.0 XML_3.99-0.16.1 lifecycle_1.0.4
[7] processx_3.8.4 lattice_0.22-6 vroom_1.6.5
[10] MASS_7.3-60.2 base64_2.0.1 scrime_1.3.5
[13] magrittr_2.0.3 limma_3.60.3 yaml_2.3.8
[16] remotes_2.5.0 httpuv_1.6.15 doRNG_1.8.6
[19] askpass_1.2.0 sessioninfo_1.2.2 pkgbuild_1.4.4
[22] DBI_1.2.3 RColorBrewer_1.1-3 abind_1.4-5
[25] pkgload_1.4.0 zlibbioc_1.50.0 quadprog_1.5-8
[28] RCurl_1.98-1.14 GenomeInfoDbData_1.2.12 genefilter_1.86.0
[31] annotate_1.82.0 DelayedMatrixStats_1.26.0 codetools_0.2-20
[34] DelayedArray_0.30.1 xml2_1.3.6 tidyselect_1.2.1
[37] UCSC.utils_1.0.0 beanplot_1.3.1 illuminaio_0.46.0
[40] GenomicAlignments_1.40.0 jsonlite_1.8.8 multtest_2.60.0
[43] ellipsis_0.3.2 survival_3.7-0 tools_4.4.0
[46] Rcpp_1.0.12 glue_1.7.0 SparseArray_1.4.8
[49] HDF5Array_1.32.0 withr_3.0.0 fastmap_1.2.0
[52] rhdf5filters_1.16.0 fansi_1.0.6 openssl_2.2.0
[55] callr_3.7.6 digest_0.6.35 timechange_0.3.0
[58] R6_2.5.1 mime_0.12 colorspace_2.1-0
[61] RSQLite_2.3.7 utf8_1.2.4 generics_0.1.3
[64] data.table_1.15.4 httr_1.4.7 htmlwidgets_1.6.4
[67] S4Arrays_1.4.1 pkgconfig_2.0.3 gtable_0.3.5
[70] blob_1.2.4 siggenes_1.78.0 htmltools_0.5.8.1
[73] profvis_0.3.8 scales_1.3.0 png_0.1-8
[76] rstudioapi_0.16.0 reshape2_1.4.4 tzdb_0.4.0
[79] rjson_0.2.21 nlme_3.1-164 curl_5.2.1
[82] cachem_1.1.0 rhdf5_2.48.0 miniUI_0.1.1.1
[85] restfulr_0.0.15 desc_1.4.3 GEOquery_2.72.0
[88] pillar_1.9.0 grid_4.4.0 reshape_0.8.9
[91] vctrs_0.6.5 urlchecker_1.0.1 promises_1.3.0
[94] xtable_1.8-4 cli_3.6.1 compiler_4.4.0
[97] Rsamtools_2.20.0 rlang_1.1.3 crayon_1.5.3
[100] rngtools_1.5.2 nor1mix_1.3-3 mclust_6.1.1
[103] ps_1.7.6 plyr_1.8.9 fs_1.6.4
[106] stringi_1.8.4 BiocParallel_1.38.0 htm2txt_2.2.2
[109] munsell_0.5.1 Matrix_1.7-0 hms_1.1.3
[112] sparseMatrixStats_1.16.0 bit64_4.0.5 Rhdf5lib_1.26.0
[115] KEGGREST_1.44.1 statmod_1.5.0 shiny_1.8.1.1
[118] memoise_2.0.1 bit_4.0.5