
Hello, I've been having a fair amount of trouble trying to create a dba object in diffbind from a .csv samplesheet. I used this code before, but I'm reanalyzing some data and I keep getting the error :
"Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec, : line 1 did not have 10 elements"
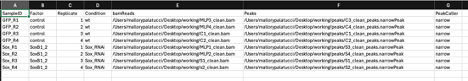
I'm not sure why it is expecting 10 columns. I checked the format to make sure my delimiters were correct and tried specifying sep = ",". I'm using narrowpeak format from MACS2 output. I also tripled checked my directories for my files to ensure my paths are accurate. Please see my code below along with the screenshot of my samplesheet attached. Any help would be greatly appreciated.
ATAC <- read.csv("ATACSox_samplesheet2023_Genrich05.csv")
SoxData <- dba(sampleSheet = ATAC)
sessioninfo() R version 4.4.0 (2024-04-24) Platform: aarch64-apple-darwin20 Running under: macOS Sonoma 14.5
Matrix products: default BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale: 1 en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: America/Los_Angeles tzcode source: internal
attached base packages: 1 stats4 stats graphics grDevices utils datasets methods base
other attached packages:
1 lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2 readr_2.1.5 tidyr_1.3.1
[8] tibble_3.2.1 ggplot2_3.5.1 tidyverse_2.0.0 DiffBind_3.14.0 SummarizedExperiment_1.34.0 Biobase_2.64.0 MatrixGenerics_1.16.0
[15] matrixStats_1.3.0 GenomicRanges_1.56.0 GenomeInfoDb_1.40.1 IRanges_2.38.0 S4Vectors_0.42.0 BiocGenerics_0.50.0
loaded via a namespace (and not attached):
1 bitops_1.0-7 deldir_2.0-4 rlang_1.1.4 magrittr_2.0.3 compiler_4.4.0 png_0.1-8 vctrs_0.6.5 pwalign_1.0.0
[9] pkgconfig_2.0.3 crayon_1.5.2 fastmap_1.2.0 XVector_0.44.0 caTools_1.18.2 utf8_1.2.4 Rsamtools_2.20.0 rmarkdown_2.27
[17] tzdb_0.4.0 UCSC.utils_1.0.0 xfun_0.44 zlibbioc_1.50.0 jsonlite_1.8.8 DelayedArray_0.30.1 BiocParallel_1.38.0 jpeg_0.1-10
[25] irlba_2.3.5.1 parallel_4.4.0 R6_2.5.1 stringi_1.8.4 RColorBrewer_1.1-3 rtracklayer_1.64.0 SQUAREM_2021.1 limma_3.60.2
[33] numDeriv_2016.8-1.1 Rcpp_1.0.12 knitr_1.47 timechange_0.3.0 Matrix_1.7-0 tidyselect_1.2.1 rstudioapi_0.16.0 abind_1.4-5
[41] yaml_2.3.8 gplots_3.1.3.1 codetools_0.2-20 curl_5.2.1 hwriter_1.3.2.1 lattice_0.22-6 plyr_1.8.9 withr_3.0.0
[49] ShortRead_1.62.0 coda_0.19-4.1 evaluate_0.24.0 Biostrings_2.72.1 pillar_1.9.0 KernSmooth_2.23-24 generics_0.1.3 RCurl_1.98-1.14
[57] invgamma_1.1 truncnorm_1.0-9 emdbook_1.3.13 hms_1.1.3 munsell_0.5.1 scales_1.3.0 ashr_2.2-63 gtools_3.9.5
[65] glue_1.7.0 tools_4.4.0 apeglm_1.26.0 interp_1.1-6 BiocIO_1.14.0 BSgenome_1.72.0 locfit_1.5-9.9 GenomicAlignments_1.40.0
[73] systemPipeR_2.10.0 XML_3.99-0.16.1 mvtnorm_1.2-5 grid_4.4.0 bbmle_1.0.25.1 amap_0.8-19 bdsmatrix_1.3-7 latticeExtra_0.6-30
[81] colorspace_2.1-0 GenomeInfoDbData_1.2.12 restfulr_0.0.15 cli_3.6.2 GreyListChIP_1.36.0 fansi_1.0.6 mixsqp_0.3-54 S4Arrays_1.4.1
[89] gtable_0.3.5 digest_0.6.35 SparseArray_1.4.8 ggrepel_0.9.5 rjson_0.2.21 htmlwidgets_1.6.4 htmltools_0.5.8.1 lifecycle_1.0.4
[97] httr_1.4.7 statmod_1.5.0 MASS_7.3-61

Thank you for your reply! There mustve been something wierd going on with my peak formats as you pointed out. I recalled all the peaks and it worked.