
Hi there. I need a sounding board and some technical advice regarding voomaByGroup().
I have a bulk RNAseq data set of cells cultured in vitro under various conditions. (The experimental design is reasonably complex with different culture conditions, patient-derived cells and such.)
There is cell type 1 and cell type 2. For cell type 1, there are two different subtypes, a and b. Cell types 1 and 2 are either cultured together or alone.
I am interested in differential gene expression between 1a and 1b. (It's explorative)
Looking at unsupervised clustering, there are two clusters related to the presence of cell type 2. One cluster is cell type 1 only (orange), and the other is cell type 1+2 or only 2 (blue).
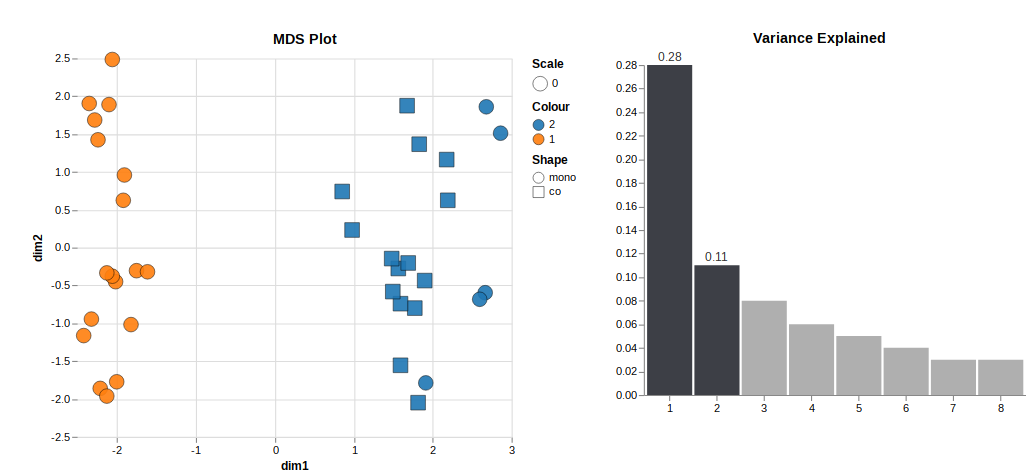
To me, that indicates that the presence of cell type 2 contributes a lot to the variance in the data set. It makes sense because it's a different cell type. But it also makes it challenging to analyse the data because I am mainly interested in the differentially expressed genes in subgroups within blue and orange.
Using lmFit() and eBayes(), there are barely any DEGs. I have tried to use voomWithQualityWeights() but it didn't help. I don't think that the overall heterogeneity is the challenge but more the two groups that arise. As I understand, voomaByGroup() might be suitable for this scenario, but I can't seem to get it to work.
This is my problem with voomaByGroup():
va <-
voomaByGroup(
y,
design = design,
group = groups
)
I tried my actual data and my actual grouping vector:
Error in sx[, lev] <- voomi$meanvar.trend$x :
number of items to replace is not a multiple of replacement length
As I understand group is a vector that indicates the group membership of the columns of y. So, the length(groups) should be equal to ncol(y), correct? After getting the error, I just assigned some random groups, to see if maybe my grouping variable is off. I get the same error if length(groups) == ncol(y), no matter how I do the grouping. I tried around if it's maybe ncol(y) - 1 or +1. But that didn't help either (kinda obviously...). What am I missing here?
Apart from the technical difficulties, according to a reply here, voomaByGroup() is quite similar to subsetting the data (which I could also do instead). Of course, as the researcher, I should know if it's reasonable to subset the data. I might just need a sounding board for this decision. In my understanding (and I am quite new to the sequencing world), having all samples together would mean more residual degrees of freedom and, ultimately, greater statistical power. However, separating the data sets might be beneficial if the subsets are indeed different, which, looking at the unsupervised clustering, is the case. Biologically, these are 2 different cell types with different gene expression patterns.
I have searched for similar questions and there seems to be no one-size-fits-all solution. As usual in research.
- what causes the difference in the topTable results by the two analyses using limma?
- Limma DE analysis using all microarray or subset of interest microarray
- limma, analysis on subset of data gives completely different results
- Limma analysis on a subset of data
Any guidance for the voomaByGroup() function or any advice/discussion around subsetting the data is greatly appreciated.
Many thanks! - Anna
