
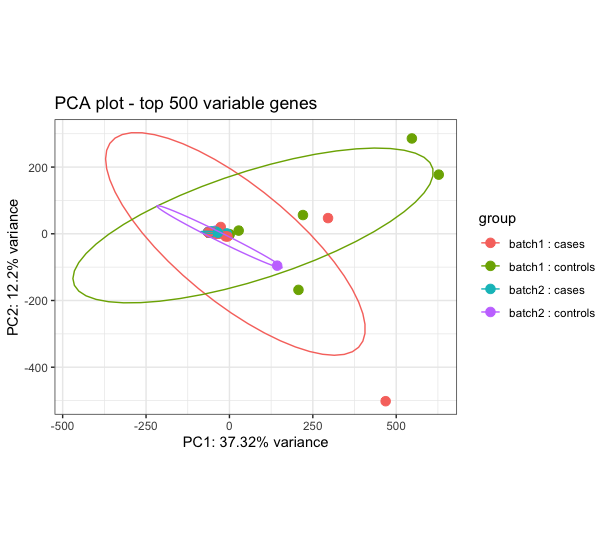
I am determining differential transcript expression and usage between cases (dually infected) and controls (single infection). I have 59 sequences with 36 cases and 23 controls. The DTE results show some signal relevant to the conditions in question, however I am constrained to trust the results because the cases and controls largely overlap on the PCA performed using rlog transformed counts (see image attached). I have performed robust PCA to try and identify outliers but it doesn't help and also used surrogate variable analysis (SVA) and combat_seq to try to adjust for hidden batch effect, but none has improved my the PCA. The challenge is pronounced when I attempt to determine Isoform switch analysis using IsoformSwitchAnalyzeR, there is no isoform switching between cases and controls. When I use visual inspection and just select 8 cases and 8 controls I get better performance and even get isoform switches. How can I best handle this issue of overlapping cases and controls?

 The seven samples have a red bar.
The seven samples have a red bar.
I think I am about to run into the same issue with my data, so I'll be interested to read peoples opinions on this also i.e. when the groups don't separate no matter what you do. I have a few ideas based on the fact that the 2-dimensional PCA is only accounting for ~ 50% of the variance in 500 genes
Thanks BioinfGuru
So as of now, I am still nearly at square one.
No worries, wish I could be more help. Plotly provide a 3-D plot function that's pretty simple (the two you did are still 2-D plots, they just differ in which 2 PCs are plotted) but I doubt it will make any difference here.
Just 2 more ideas:
Did you make any headway?
Have you tried plotting a hierarchical clustering heatmap on tpm normalised count data without any adjustment for hidden variables (combat-seq, SVA etc) ? It is simple and fast. Just to see if any samples/groups cluster at all on the original data. It might be that you are removing too much biological signal.