
Hello. I'm using edgeR to examine how gene expression changes in a control cell line vs. a genetically modified cell line at three timepoints. Each condition (ex. CTRL_DIV2, MOD_DIV2) has 3 replicates for a total of 18 samples.
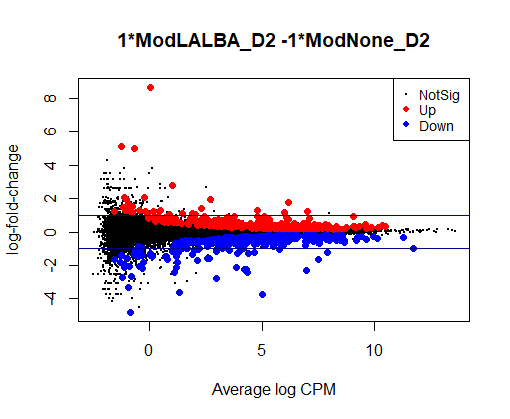
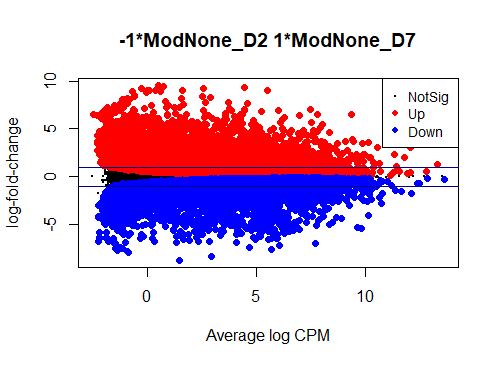
I see typical small DE gene ratios for contrasts between Mod - CTRL at the same timepoint, but a HUGE amount of DE genes (70 - 80%) in contrasts between different timepoints of the same cell line. Alongside timepoints, there are significant changes in cell culture conditions: D2 corresponds to 50% confluence, D4 100% confluence, and D7 is intentional overconfluence, glucose starvation and formation of complex structures.
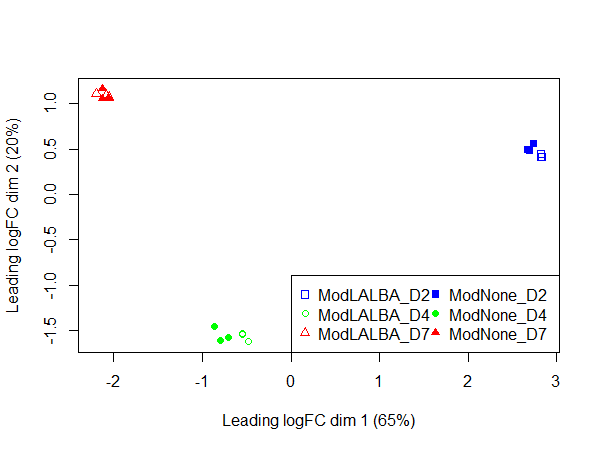
In the PCA plot, the replicates cluster well together and it appears most of the variation is explained by time in culture, consistent with the DE values.
Could anyone offer advice - could it be possible that this is biologically real?
EDIT: Cleaned up relevant code and including below:
## Prepare data files
fc <- readRDS("RNAseq.rds")
counts <- fc$counts
colnames(counts) <- sub("\\_.*", "", colnames(counts))
meta <- read.csv("metadata.csv")
meta <- meta[order(row.names(meta)),]
Group <- factor(paste0("Mod", meta$Mod, "_D", meta$DIV))
head(Group)
# [1] ModNone_D2 ModLALBA_D4 ModLALBA_D4 ModLALBA_D4 ModNone_D7
# [6] ModNone_D7
# 6 Levels: ModLALBA_D2 ModLALBA_D4 ModLALBA_D7 ... ModNone_D7
## Create DGElist
y <- DGEList(counts = counts,
group = Group,
lib.size = colSums(counts))
colnames(y) <- rownames(meta)
## Build and Ensembl-specific annotation lookup
mart <- useMart("ENSEMBL_MART_ENSEMBL")
mart <- useDataset("hsapiens_gene_ensembl", mart)
ens <- row.names(y)
ensLookup <- gsub("\\.[0-9]*$", "", ens)
head(ensLookup)
annotLookup <- getBM(
mart=mart,
attributes=c("ensembl_transcript_id", "ensembl_gene_id",
"gene_biotype", "external_gene_name", "entrezgene_id"),
filter="ensembl_gene_id",
values=ensLookup,
uniqueRows=TRUE)
annotLookup <- data.frame(
ens[match(annotLookup$ensembl_gene_id, ensLookup)],
annotLookup)
colnames(annotLookup) <- c("original_id", c("ensembl_transcript_id", "ensembl_gene_id", "gene_biotype", "external_gene_name", "entrezgene_id"))
summary(annotLookup)
original_id ensembl_transcript_id ensembl_gene_id
Length:259537 Length:259537 Length:259537
Class :character Class :character Class :character
Mode :character Mode :character Mode :character
gene_biotype external_gene_name entrezgene_id
Length:259537 Length:259537 Min. : 1
Class :character Class :character 1st Qu.: 7018
Mode :character Mode :character Median : 51307
Mean : 12696013
3rd Qu.: 146822
Max. :128966744
NA's :53659
## Annotation mapping
Symbol <- mapIds(org.Hs.eg.db,
keys=rownames(y),
keytype="ENSEMBL",
column="SYMBOL")
Symbol.df <- data.frame(Symbol=Symbol)
Symbol.df$ENTREZID <- annotLookup$entrezgene_id[match(Symbol.df$Symbol, annotLookup$external_gene_name)]
y$genes <- Symbol.df
## Filter genes and create design matrix
keep <- filterByExpr(y) # gene pool (including non-coding RNAs) size reduced from 62,696 --> 23,574
design <- model.matrix(~0 + Group)
colnames(design) <- levels(Group)
keep2 <- filterByExpr(y, design)
x <- y[keep2, , keep.lib.sizes = FALSE]
x <- calcNormFactors(x)
## Print MD plots in console (no QC issues detected)
#pdf(file = "MeanDifferencePlots.pdf")
#par(mfrow = c(3,3))
#for(i in 1:18){
# print((plotMD(cpm(x, log=TRUE), column=i) + abline(h=0, col="red", lty=2, lwd=2)))
#}
points <- c(0,1,2,15,16,17)
colors <- rep(c("blue", "green", "red"), 2)
plotMDS(x, col=colors[Group], pch = points[Group])
legend("topleft", legend = levels(Group), pch = points, col = colors, ncol = 2)
# Estimate dispersion
x <- estimateDisp(x, design, robust = TRUE)
x$common.dispersion
# [1] 0.003289937
plotBCV(x)
saveRDS(x, file = "processed_data.rds")
x <- readRDS("processed_data.rds")
## Fit a GLM
fit <- glmQLFit(x, design, robust = TRUE)
QLDplot <- plotQLDisp(fit)
## Differential expression for one contrast (reference = edgeR manual section 4.4.8)
con1 <- makeContrasts(ModNone_D4 - ModNone_D2, levels=design)
qlf1 <- glmQLFTest(fit, contrast=con1)
summary(decideTests(qlf1, p.value = 0.05))
# -1*ModNone_D2 1*ModNone_D4
#Down 7687
#NotSig 5743
#Up 7906
#integer(0)
plotMD(qlf1) + abline(h = c(-1,1), col = "darkblue")
qlf1.df <- as.data.frame(qlf1)
EnhancedVolcano(qlf1.df,
x = "logFC",
y = "PValue",
lab = qlf1.df$Symbol,
FCcutoff = 1,
pCutoff = 10e-6,
title = "P2D11 NV DIV4 - P2D11 NV DIV2")
## Output tables for top DE genes and pathways
go <- goana(qlf1, species = "Hs", geneid = "ENTREZID")
topGOup <- topGO(go, ontology = "BP", sort = "up", n=100)
topGOdown <- topGO(go, ontology = "BP", sort = "down", n=100)
top1 <- as.data.frame(topTags(qlf1, n = 1000))
top1 <- top1[top1$FDR < 0.05,]
top1up <- top1[top1$logFC > 0,]
top1down <- top1[top1$logFC < 0,]
sessionInfo()
```
R version 4.2.2 (2022-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 22631)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8
[2] LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8
[4] LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets
[7] methods base
other attached packages:
[1] EnhancedVolcano_1.16.0 ggrepel_0.9.4
[3] knitr_1.43 ggplot2_3.4.4
[5] dplyr_1.1.2 plyr_1.8.8
[7] openxlsx_4.2.5.2 GO.db_3.16.0
[9] biomaRt_2.54.1 org.Hs.eg.db_3.16.0
[11] AnnotationDbi_1.60.2 IRanges_2.32.0
[13] S4Vectors_0.36.2 Biobase_2.58.0
[15] BiocGenerics_0.44.0 Rsubread_2.12.3
[17] edgeR_3.40.2 limma_3.54.2
loaded via a namespace (and not attached):
[1] httr_1.4.7 splines_4.2.2
[3] bit64_4.0.5 statmod_1.5.0
[5] BiocManager_1.30.22 BiocFileCache_2.6.1
[7] blob_1.2.4 GenomeInfoDbData_1.2.9
[9] yaml_2.3.7 progress_1.2.2
[11] pillar_1.9.0 RSQLite_2.3.1
[13] lattice_0.20-45 glue_1.6.2
[15] digest_0.6.31 RColorBrewer_1.1-3
[17] XVector_0.38.0 colorspace_2.1-0
[19] htmltools_0.5.6 Matrix_1.6-1
[21] XML_3.99-0.14 pkgconfig_2.0.3
[23] zlibbioc_1.44.0 purrr_1.0.2
[25] scales_1.2.1 tibble_3.2.1
[27] KEGGREST_1.38.0 farver_2.1.1
[29] generics_0.1.3 cachem_1.0.8
[31] withr_2.5.0 cli_3.6.1
[33] magrittr_2.0.3 crayon_1.5.2
[35] memoise_2.0.1 evaluate_0.21
[37] fansi_1.0.4 xml2_1.3.5
[39] textshaping_0.3.6 tools_4.2.2
[41] prettyunits_1.1.1 hms_1.1.3
[43] lifecycle_1.0.3 stringr_1.5.0
[45] munsell_0.5.0 locfit_1.5-9.8
[47] zip_2.3.0 Biostrings_2.66.0
[49] compiler_4.2.2 GenomeInfoDb_1.34.9
[51] systemfonts_1.0.4 rlang_1.1.1
[53] grid_4.2.2 RCurl_1.98-1.12
[55] rstudioapi_0.15.0 rappdirs_0.3.3
[57] labeling_0.4.2 bitops_1.0-7
[59] rmarkdown_2.24 gtable_0.3.3
[61] DBI_1.1.3 curl_5.1.0
[63] R6_2.5.1 fastmap_1.1.1
[65] bit_4.0.5 utf8_1.2.3
[67] filelock_1.0.2 ragg_1.2.5
[69] stringi_1.7.12 Rcpp_1.0.11
[71] vctrs_0.6.3 png_0.1-8
[73] dbplyr_2.3.3 tidyselect_1.2.0
[75] xfun_0.40
```

Hi Gordon, thank you for replying! I've updated the original post to include code.
The NG dispersion is almost zero for your data, meaning that there is almost no biological variation between the replicates. There are also heaps of genes with very, very large fold-changes between the timepoints. So it's inevitable you'll get very large numbers of DE genes.
I'd recommend you use glmTreat() to reduce the number of DE genes before doing a GO analysis.
You might consider upgrading to the current version of Bioconductor, although it won't make much difference to your current analysis.