Entering edit mode

I am trying to subset a df of normalized counts and then make a heatmap using pheatmap() (essentially subsetting the heatmaps), but am running into the NA/NaN/Inf error. Interestingly, I am successfully able to create a heatmap with the non-subsetted normalized counts, but once I subset it, I am unable to generate the heatmap. Below is a snippet of my code. Thanks!
Also, I subsetted the df_normalized_counts_2 (rowxcol = genexsample) by the genes and didn't run into this issue (code for it not shown)
vsd_2 <- vst(dds_2, blind=FALSE) # generating a matrix of values for which variance is constant across range of mean values
df_normalized_counts_2 <- assay(vsd_2)
df_subset_3 <- df_pre_subset[df_pre_subset$condition_and_cellline %in% comparison_3,, drop = FALSE]
subsetted_samples_3 <- rownames(df_subset_3)
subsetted_samples_3
df_normalized_counts_3 <- assay(vsd_2)[,subsetted_samples_3]
options(repr.plot.width=7, repr.plot.height=5)
pheatmap(df_normalized_counts_3.2,
cluster_rows = TRUE, # Cluster rows (genes)
cluster_cols = TRUE, # Cluster columns (samples)
scale = "row", # Scale rows (genes) for better visualization
color = colorRampPalette(c("green", "black", "red"))(100), # Choose a color palette
main = "DEGs Heatmap", # Heatmap title
fontsize = 8) # Adjust fontsize for gene names
It would something like this:
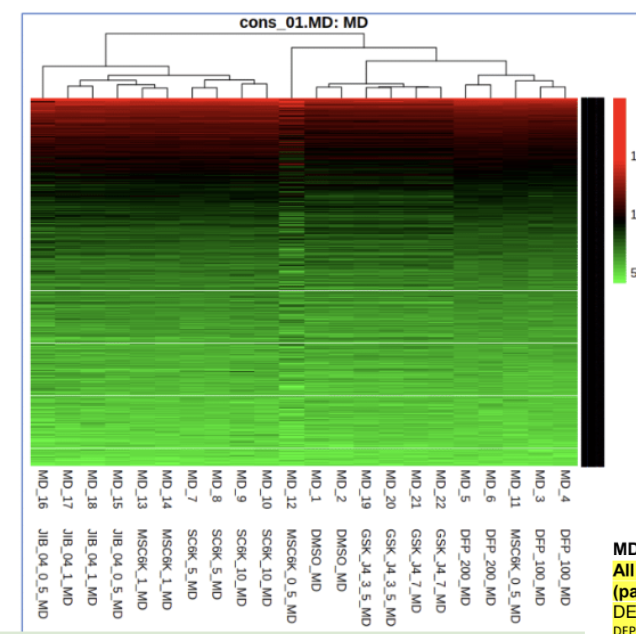
Heatmap non-subsetted:
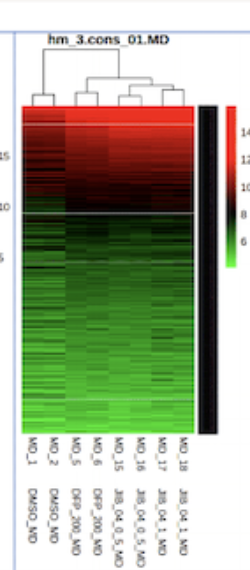
Heatmap subsetted:

Thank you for your response. To clarify, the process of making a heatmap performs rowscaling so it takes the value - (mean/std). And to resolve this, I subset out the rows that have the same values across (this worked and I'm able to generate the subsetted heatmaps!). However, wouldn't this impact the analysis by removing those genes and bias the interpretation of the heatmap?
Ask yourself the question how a gene that is exactly the same everywhere adds anything to any analysis. Heatmaps usually take DEGs to infer patterns.