
Hello,
I have a problem that I can just not wrap my head around. I am new to bioinformatics (coming from a computer science background), so I might have overlooked something really obvious.
I have a couple of genes that I need to get the 1500 base pairs upstream and the first 500 bases of (so 2000 bases in total). I then want to find motifs in those sequences. I want to do so using FIMO and MAST from the MEME suite. For this reason, I want my output to be formatted appropriately, as this facilitates downstream processing of the resulting gff3 files.
I obtained the genome and the annotation from here and here, respectively.
As for the tooling in this example, conda install agat bedtools should suffice.
Then, in approach A, I am retrieving the sequences like this:
agat_sp_filter_feature_from_keep_list.pl --gff ../reference_dbs/Homo_sapiens.GRCh38.111.gff3 --keep_list output_human/genes_trafo -a Name -o output_human/output_trafo.gff3
sed -i.old '1s;^;##seqid source type start end score strand phase attributes;' output_human/output_trafo_genesonly.gff3
agat_sp_extract_sequences.pl -g output_human/output_trafo.gff3 -t gene -f ../reference_dbs/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa --output output_human/sequences_from_paper_genes.fa
python reformat_fasta.py -i output_human/sequences_from_paper_genes.fa -o output_human/sequences_from_paper_genes_stripped.fa
agat_sp_extract_sequences.pl -g output_human/output_trafo.gff3 -t gene -f ../reference_dbs/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa --output output_human/sequences_from_paper_genes_1500up.fa --up 1500
python reformat_fasta.py -i -t 2000 output_human/sequences_from_paper_genes_1500up.fa -o output_human/sequences_from_paper_genes_1500up_500down.fa
where for the purposes of just a minimal example, output_human/genes_trafo just contains
ABCA9
ABLIM3
ABTB2
ACSL1
ADAMDEC1
ADAR
ADM
AHNAK2
AIM2
AKT3
A short check with ensemble (which appears to be non-functional at the moment) suggested that those sequences are correct. The problem is that the header is not formatted properly, so the output from MEME would not contain absolute coordinates, making downstream processing much harder.
When searching for a tool that produces the desired headers, I found that bedtools getfasta produces the correct headers.
So, in a second approach, I tried retrieving the sequences using bedtools:
grep "ID=gene" output_human/output_trafo.gff3 > output_human/output_trafo_genesonly.gff3
gff2bed < output_human/output_trafo_genesonly.gff3 > output_human/output_trafo_genesonly.bed
bedtools getfasta -bed output_human/output_trafo_genesonly.bed -fi ../reference_dbs/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa > output_human/sequences_from_paper_genes_ucsc.fa
bedtools flank -l 1500 -r 0 -i output_human/output_trafo_genesonly.bed -g ../reference_dbs/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa.fai > output_human/flank.bed
bedtools getfasta -bed output_human/flank.bed -fi ../reference_dbs/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa > output_human/flank.fa
python3 merge_fasta_linebyline.py -i output_human/flank.fa output_human/sequences_from_paper_genes_ucsc.fa -o output_human/sequences_from_paper_genes_ucsc_1500up.fa
python reformat_fasta.py -t 2000 -i output_human/sequences_from_paper_genes_ucsc_1500up.fa -o output_human/sequences_from_paper_genes_ucsc_1500up500down.fa
But I found that, while the resulting gff3 and bed files are compatible, the files sequences_from_paper_genes.fa (approach A) and sequences_from_paper_genes_ucsc.fa (approach B) are different:
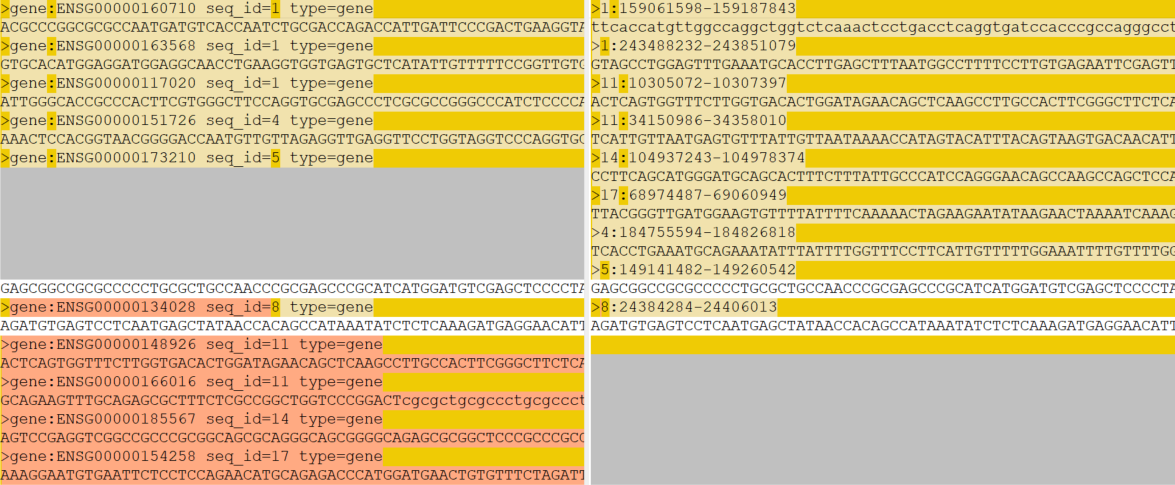
The scripts reformat_fasta.py and merge_fasta_linebyline.py should be fine, but I'm providing them here anyway (not my finest pieces of engineering, but they do the job):
## merge_fasta_linebyline.py
@cloup.command()
@cloup.option("--input-fasta", "-i", nargs=2, type=cloup.File("r"), required=True, help="input fasta file")
@cloup.option("--output-fasta", "-o", type=cloup.File("w"), required=True, help="input fasta file")
def main(input_fasta, output_fasta):
in1, in2 = input_fasta
for line1, line2 in zip(in1, in2):
if line1.startswith(">"):
assert (line2.startswith(">"))
rgx = r"\>(.*):(\d+)\-(\d+)"
m1 = re.match(rgx, line1)
m2 = re.match(rgx, line2)
assert m1.group(1) == m2.group(1)
chr = m1.group(1)
range1 = (int(m1.group(2)), int(m1.group(3)))
range2 = (int(m2.group(2)), int(m2.group(3)))
assert (range1[1] == range2[0])
assert (range1[0] <= range2[1])
outrange = (range1[0], range2[1])
output_fasta.write(f">{chr}:{outrange[0]}-{outrange[1]}\n")
else:
# remove trailing newline (to avoid splitting the sequence!)
output_fasta.write(line1.strip()+line2)
## reformat_fasta.py
@cloup.command()
@cloup.option("--input-fasta", "-i", type=cloup.File("r"), required=True, help="input fasta file")#
@cloup.option("--output-fasta", "-o", type=cloup.File("w"), required=True, help="input fasta file")
@cloup.option("--trim-sequences", "-t", type=int, required=False, help="input fasta file")
def main(input_fasta, output_fasta, trim_sequences):
outline = ""
for line in input_fasta:
if line.startswith(">"):
if outline:
if trim_sequences:
outline = outline[:trim_sequences]
output_fasta.write(f"{outline}\n")
outline = ""
output_fasta.write(f"{line}")
else:
outline += line.strip()
if outline:
output_fasta.write(f"{outline}")
I'm puzzled that there seems to be no systematic error, as two of the sequences are completely identical, but the rest looks like rubbish to me. Does anybody have an idea what's going on or how I could debug this further? Please feel free to add appropriate tags to the question (I'd suggest agat and bedtools but apparently I lack rights to create those tags)
Hi, in the future please post at e.g. biostars.org as this is not related to Bioconductor.
The common ones are the genes without UTRs while the different ones are gene having UTRs, so I guess AGAT and bedtools (gff2bed step?) do not handle the same way the UTRs. UTRs are part of the gene, so the upstream region should be upstream of the UTR, could you check a gene that is different manually on Ensembl to see what region is extracted (you can use blat if it is too complicated to check manually)