Entering edit mode

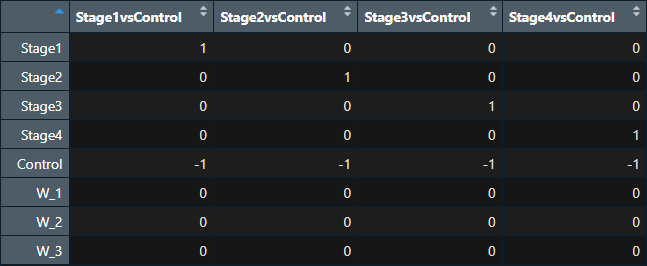
I have 3 estimated W (factors of unwanted variation) after RUVg correction. I am trying to include W (estimated factors of unwanted variation) into the design matrix just as recommended by Prof. Davide Risso [2] but I am concerned about how this will affect the contrast matrix? They appear as 3 extra rows compared to the desired contrast matrix (to have 5 rows and 5 columns). Is there some wrong application here? W is a matrix of 3 columns (W_1, W_2, W_3)
design=model.matrix(~0+W+dge$samples$Stage)
colnames(design)[1] ="W_1"
colnames(design)[2] ="W_2"
colnames(design)[3] ="W_3"
colnames(design)[4] ="Control"
colnames(design)[5] ="Stage1"
colnames(design)[6] ="Stage2"
colnames(design)[7] ="Stage3"
colnames(design)[8] ="Stage4"
###Change columns order###
design <- design[, c(5,6,7,8,4,1,2,3)]
v = voomWithQualityWeights(dge, design = design, plot = TRUE)
vfit <- lmFit(v, design)
contrast.matrix <- makeContrasts(Stage1vsControl=Stage1-Control,
Stage2vsControl=Stage2-Control,
Stage3vsControl=Stage3-Control,
Stage4vsControl=Stage4-Control,
levels = colnames(design))
vfit <- contrasts.fit(vfit, contrasts=contrast.matrix)
efit <- eBayes(vfit)
plotSA(efit,main = "Final model: Mean-variance trend")
summary(decideTests(efit, method = "separate", adjust.method = "BH", p.value= 0.05,lfc = 1))

Thanks for clarifying