
Hello,
I am trying to use featureCounts() to assign RNA-seq reads to the set of annotated transcript isoforms with which they are completely consistent. When outputting this assignment to the "CORE" format (tab-separated table with columns detailing the assignment of each individual sequencing read), I found a weird bug. Occasionally, featureCounts would assign reads to the identical set of isoforms, but would output different comma-separated target list strings. For example, I had a read which was assigned to "CHS.46558.6,CHS.46558.5,CHS.46558.2,CHS.46558.11,CHS.46558.1,CHS.46558.16" and another read assigned to "CHS.46558.6,CHS.46558.5,CHS.46558.2,CHS.46558.16,CHS.46558.11,CHS.46558.1". Notice that the isoform content is identical in these two cases, its just the order they are listed in the string that differs. I created a reproducible example where I took 100 reads from the bam file with one of these two target lists and 100 with the other target list. Notably, when running featureCounts as detailed below on this downsampled bam file, the reads get assigned to the same exact target list as in the full bam file. I am hosting the data necessary to run this reproducible example, as well as the output .featureCounts file from this example, on a personal, public github repo.
### Load dependencies
library(Rsubread)
library(data.table)
### Paths to run example
samfile_path <- "path/to/example.sam"
gtf_path <- "path/to/mini_annotation.gtf"
output_dir <- "directory/to/output/CORE/file/"
### Run FeatureCounts, assigning reads to set of transcript IDs with which they are completely consistent
featureCounts(file = samfile_path,
annot.ext = gtf_path,
isGTFAnnotationFile = TRUE,
strandSpecific = 1,
isPairedEnd = TRUE,
countReadPairs = TRUE,
nonOverlap = 0,
reportReads = "CORE",
reportReadsPath = output_dir,
nthreads = 4,
primaryOnly = TRUE,
allowMultiOverlap = TRUE,
GTF.featureType = "exon",
GTF.attrType = "transcript_id")
### Assess results
core_file <- list.files(path = output_dir,
pattern = "featureCounts",
full.names = TRUE)[1]
results <- fread(core_file,
sep = "\t")
# Print counts for different target lists
results[,.N, by = V4]
V4 N
1: CHS.46558.6,CHS.46558.5,CHS.46558.2,CHS.46558.11,CHS.46558.1,CHS.46558.16 100
2: CHS.46558.6,CHS.46558.5,CHS.46558.2,CHS.46558.16,CHS.46558.11,CHS.46558.1 100
### Get session information
sessionInfo( )
R version 4.3.1 (2023-06-16 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 11 x64 (build 22621)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.utf8 LC_CTYPE=English_United States.utf8
[3] LC_MONETARY=English_United States.utf8 LC_NUMERIC=C
[5] LC_TIME=English_United States.utf8
time zone: America/New_York
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] data.table_1.14.10 Rsubread_2.16.0
loaded via a namespace (and not attached):
[1] tidyselect_1.2.0 dplyr_1.1.4
[3] blob_1.2.4 filelock_1.0.3
[5] Biostrings_2.70.1 bitops_1.0-7
[7] fastmap_1.1.1 RCurl_1.98-1.13
[9] BiocFileCache_2.10.1 GenomicAlignments_1.38.0
[11] promises_1.2.1 XML_3.99-0.15
[13] digest_0.6.34 mime_0.12
[15] lifecycle_1.0.4 ellipsis_0.3.2
[17] KEGGREST_1.42.0 RSQLite_2.3.5
[19] magrittr_2.0.3 compiler_4.3.1
[21] rlang_1.1.2 progress_1.2.3
[23] tools_4.3.1 utf8_1.2.4
[25] yaml_2.3.7 rtracklayer_1.62.0
[27] htmlwidgets_1.6.4 prettyunits_1.2.0
[29] S4Arrays_1.2.0 pkgbuild_1.4.3
[31] bit_4.0.5 curl_5.2.0
[33] DelayedArray_0.28.0 xml2_1.3.6
[35] pkgload_1.3.4 abind_1.4-5
[37] BiocParallel_1.36.0 miniUI_0.1.1.1
[39] purrr_1.0.2 BiocGenerics_0.48.1
[41] grid_4.3.1 stats4_4.3.1
[43] fansi_1.0.5 urlchecker_1.0.1
[45] profvis_0.3.8 xtable_1.8-4
[47] MASS_7.3-60 biomaRt_2.58.0
[49] SummarizedExperiment_1.32.0 cli_3.6.1
[51] crayon_1.5.2 remotes_2.4.2.1
[53] generics_0.1.3 rstudioapi_0.15.0
[55] httr_1.4.7 rjson_0.2.21
[57] sessioninfo_1.2.2 DBI_1.2.1
[59] cachem_1.0.8 stringr_1.5.1
[61] zlibbioc_1.48.0 parallel_4.3.1
[63] AnnotationDbi_1.64.1 XVector_0.42.0
[65] restfulr_0.0.15 matrixStats_1.1.0
[67] vctrs_0.6.4 devtools_2.4.5
[69] Matrix_1.6-3 IRanges_2.36.0
[71] hms_1.1.3 S4Vectors_0.40.1
[73] bit64_4.0.5 glue_1.6.2
[75] codetools_0.2-19 stringi_1.8.1
[77] later_1.3.2 GenomeInfoDb_1.38.5
[79] BiocIO_1.12.0 GenomicRanges_1.54.1
[81] tibble_3.2.1 pillar_1.9.0
[83] rappdirs_0.3.3 htmltools_0.5.7
[85] GenomeInfoDbData_1.2.11 R6_2.5.1
[87] dbplyr_2.4.0 shiny_1.8.0
[89] lattice_0.22-5 Biobase_2.62.0
[91] png_0.1-8 Rsamtools_2.18.0
[93] memoise_2.0.1 httpuv_1.6.13
[95] Rcpp_1.0.12 SparseArray_1.2.2
[97] usethis_2.2.2 MatrixGenerics_1.14.0
[99] fs_1.6.3 pkgconfig_2.0.3
Other notes:
1) I originally discovered this error using the command line interface through the featureCounts Snakemake wrapper. That's just to say the bug is not specific to the R-interface. 2) It is an oddly infrequent error. Most instances where a set of reads is consistent with several annotated isoforms yields a single unique target list. In addition, in the case I used for the reproducible example, there is a pretty large imbalance in terms of reads getting assigned to one target list or the other for the gene I saw this quirk at. Assigning reads from the full bam file yielded 18,827 reads getting assigned to "CHS.46558.6,CHS.46558.5,CHS.46558.2,CHS.46558.16,CHS.46558.11,CHS.46558.1" and only 3889 getting assigned to "CHS.46558.6,CHS.46558.5,CHS.46558.2,CHS.46558.11,CHS.46558.1,CHS.46558.16". 3) Below is an IGV screenshot of the alignments that were assigned to one target list vs. the other. Interestingly, there is some striking separation along the length of the genes for the two sets of reads. The data is data simulated from a pipeline I am developing which combines polyester with some custom scripts to simulate NR-seq data (TimeLapse-seq, SLAM-seq, etc.), so there are an abundance of T-to-C or G-to-A mutations in some of the simulated reads:
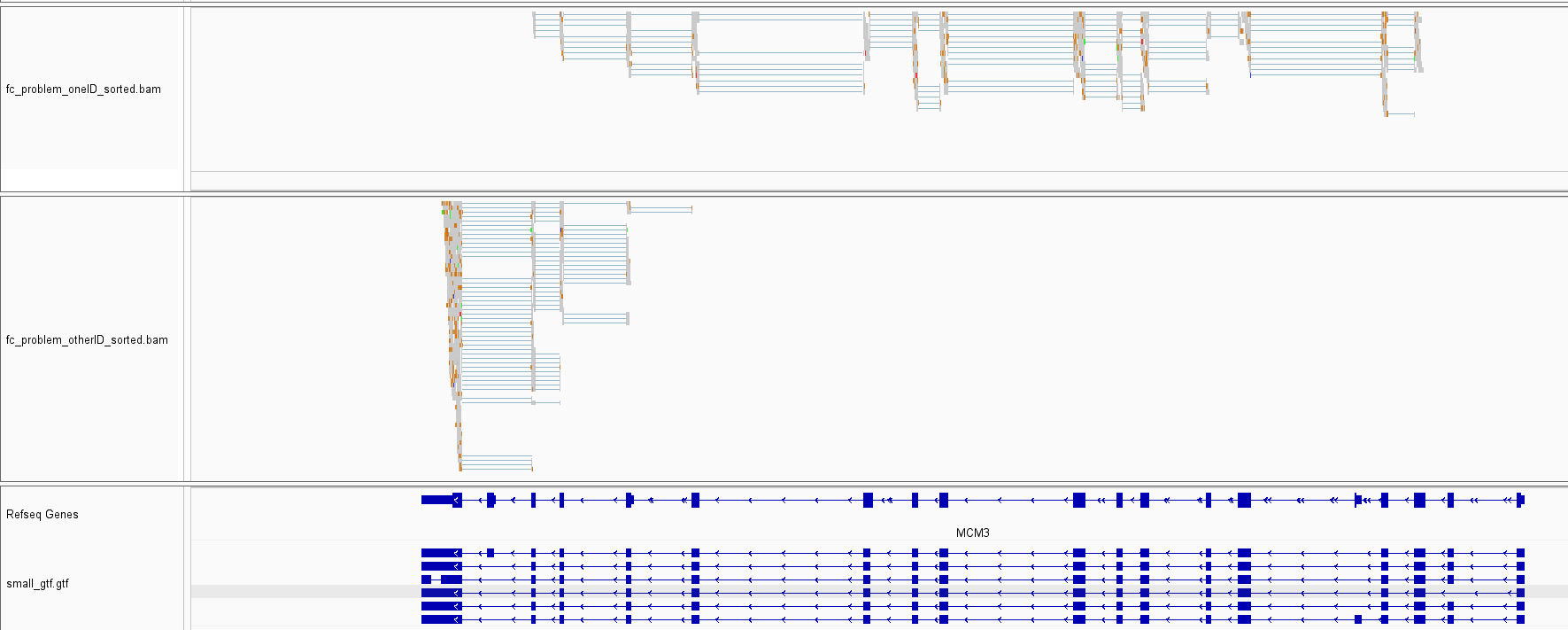
4) The quirk reproduces on both a Linux and Windows operating system.
Thank you in advance, Isaac


Thank you very much!