
Hi,
I'm using AggregateExpression() function to convert my scRNA-seq data into pseudobulk for differential expression with Deseq2. I'm wondering whether AggregateExpression() simply sums the counts for each gene in each cell, or if it also normalizes by the different numbers of cells that each sample has.
This is the Deseq2 plot for gene LRP6 showing it is differentially expressed in group MUT as compared to WT. Within the group MUT, however, I am wondering whether the sample highlighted in red has higher expression of this gene simply because it may have more cells (let's say for example 10,000), and the sample highlighted in blue has lower expression of this gene because it may have way less cells (let's say for example 500).
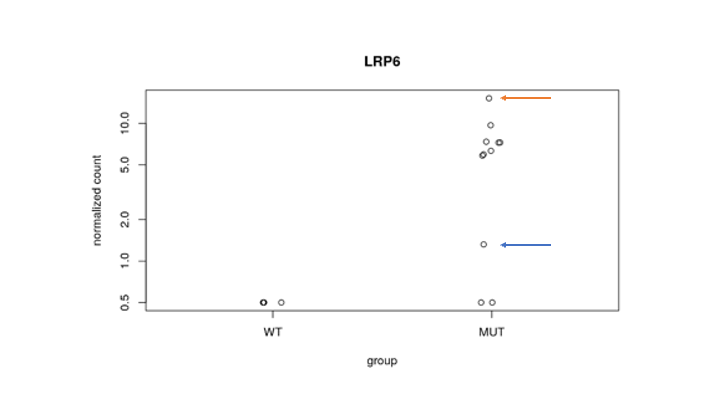
Could that be the case? And if so, how to avoid it?

Thank you. Do you have any suggestion on how to perform the normalization?
Please don't post a comment as an answer. You can refer to the OSCA book for details on normalizing pseudobulk data.
I moved it just now.
From its characteristics it's no longer sparse and similar to bulk RNA-seq so any bulk RNA-seq method should do. I personally use the normalization methods from DESeq2 and edgeR a lot.