Entering edit mode

When I set the contrast in DiffBind as
# Contrast from normalized object
dbObj_contrast_manual <- dba.contrast(dbObj_norm, design="~Condition")
dbObj_contrast_cells <- dba.contrast(dbObj_contrast_manual,group1=dbObj_norm$masks$Type1, group2=dbObj_norm$masks$Type2, name1 = "Type1", name2="Type2")
I get a log fold changes that reach up to +-1 and a reasonably looking MA-plot
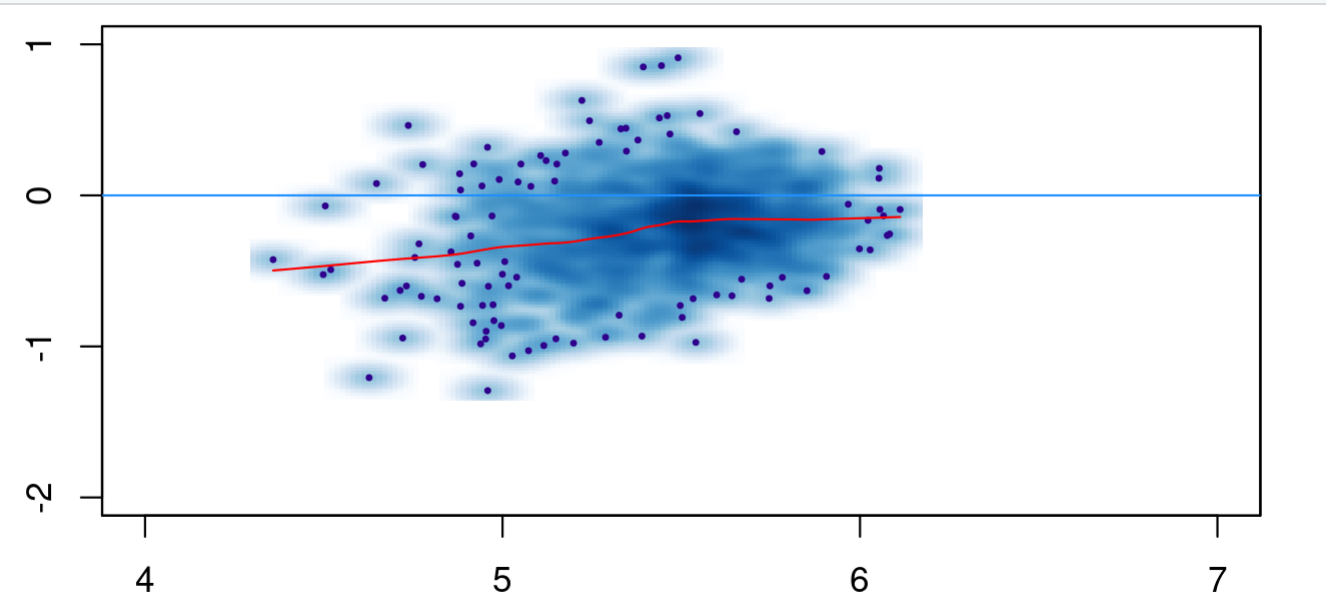
dbObj_contrast_cells <- dba.contrast(dbObj_norm,design="~Condition", contrast = c("Condition", "Type1", "Type2"))
I get fold changes near zero.
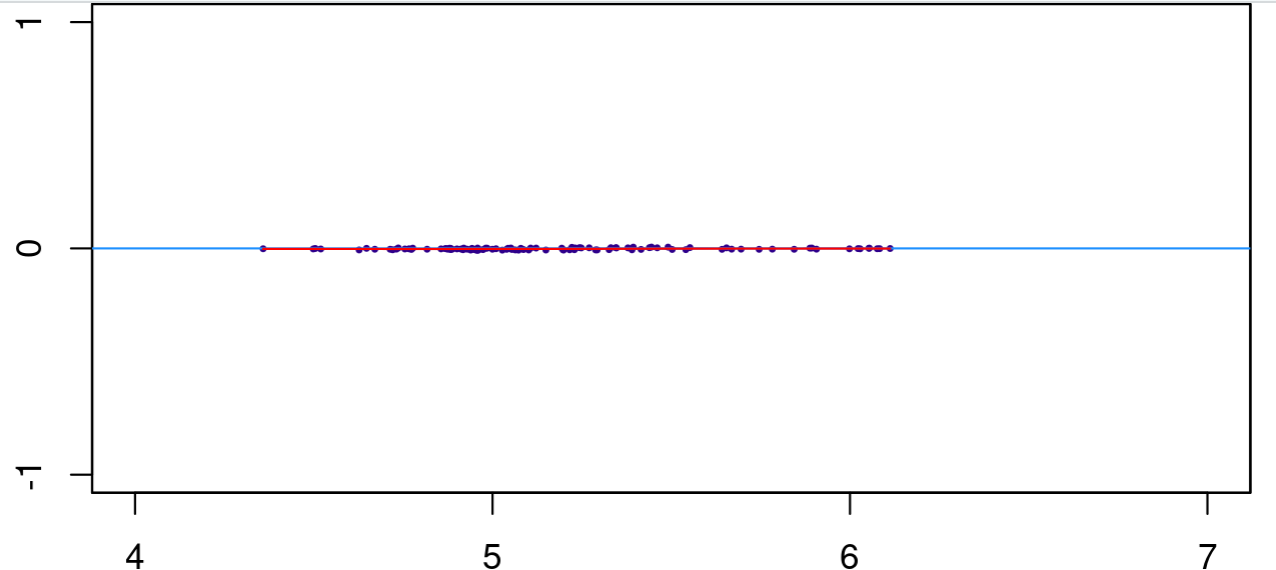
This is similar to DiffBind getting Fold values of near 0
Why is this so?
Which one is more correct?
One is the "raw" logFCs, not corrected for the standard error of the logFCs, the other one the shrunken logFCs from DESeq2 lfcShrink, "corrected" for the standard error. Essentially, if there is little evidence that the logFCs are reliable (large standard errors, low power) then they're shrunken towards zero. That is what you see. Both are "correct", but it suggests that your data are either underpowered, noisy and logFCs are not reliable. Generally, for downstream analysis I go with shrunken ones. For QC I want to see data "as-is" so I take the "raw" ones. You now need to figure out if you can do something about potential data noise. Do PCA to see if batch effects introduce unwanted variation. Maybe it can be corrected for.
lfcShrink is from DESeq2 (like you said). But don't we expect the evidence in ChIP-Seq to be (generally) weaker than the evidence in an RNA-Seq experiment (very generally speaking) ? If that is indeed the case, then I have doubts that shrinking the log fold change in the same manner as is done in RNA-Seq is appropriate here.