
Hello everyone,
I've been working extensively with a small RNA-seq dataset comprising 47 samples, each associated with a specific date and categorized into 'ON' or 'OFF' conditions, with three replicates per condition (except for one condition with two replicates only). This dataset encompasses the counts of 77,000 small RNA molecules detected on our reference genome using short stack.
I've conducted my DESeq2 analysis using the following code setup:
dds <- DESeqDataSetFromMatrix(countData = data, colData = metadata, design = ~ lab) dds <- DESeq(dds, betaPrior = TRUE)
Utilizing the results() function, I extracted differential expression results for each 'date + condition (lab)' combinations:
contrast <- c("lab", paste0(day, "ON"), paste0(day, "OFF")) res <- results(dds, contrast = contrast) result_data <- as.data.frame(res)
However, when examining the count plots, I've observed a frequent occurrence of significant adjusted p-values between conditions particularly on date 4 when it shouldn't be, this occurrence seems unexpected (example 1).
example 1 :
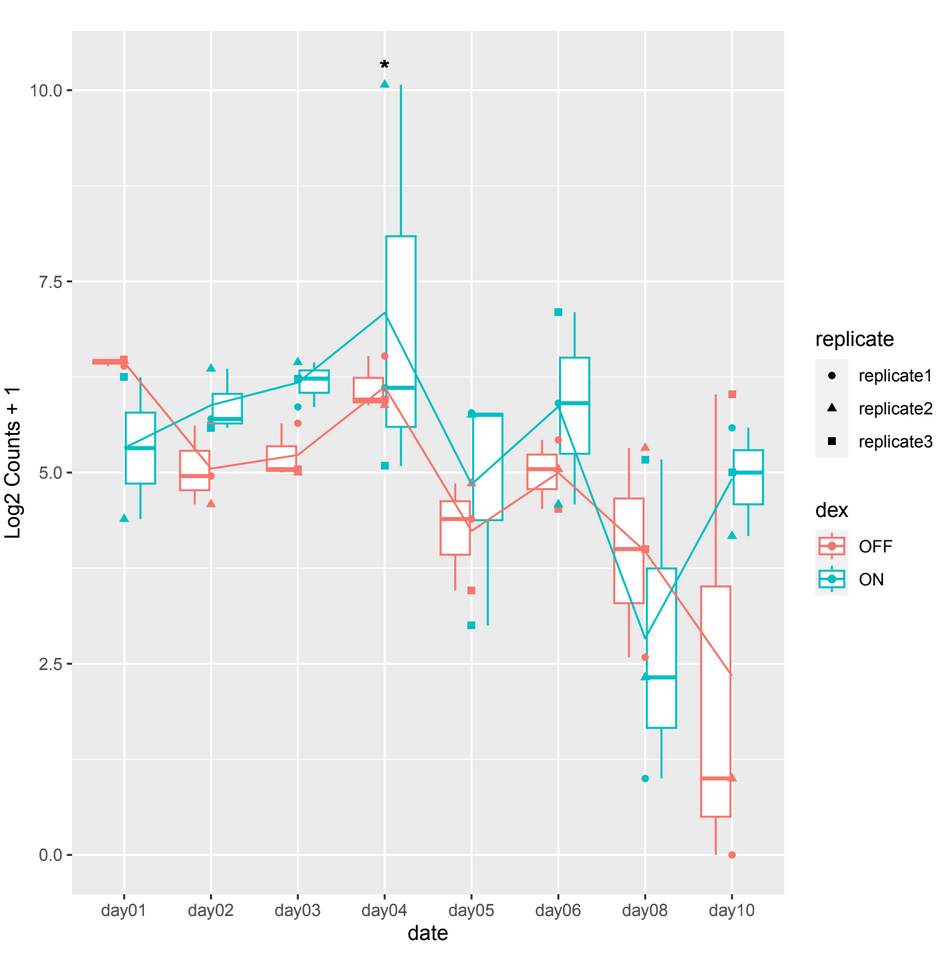
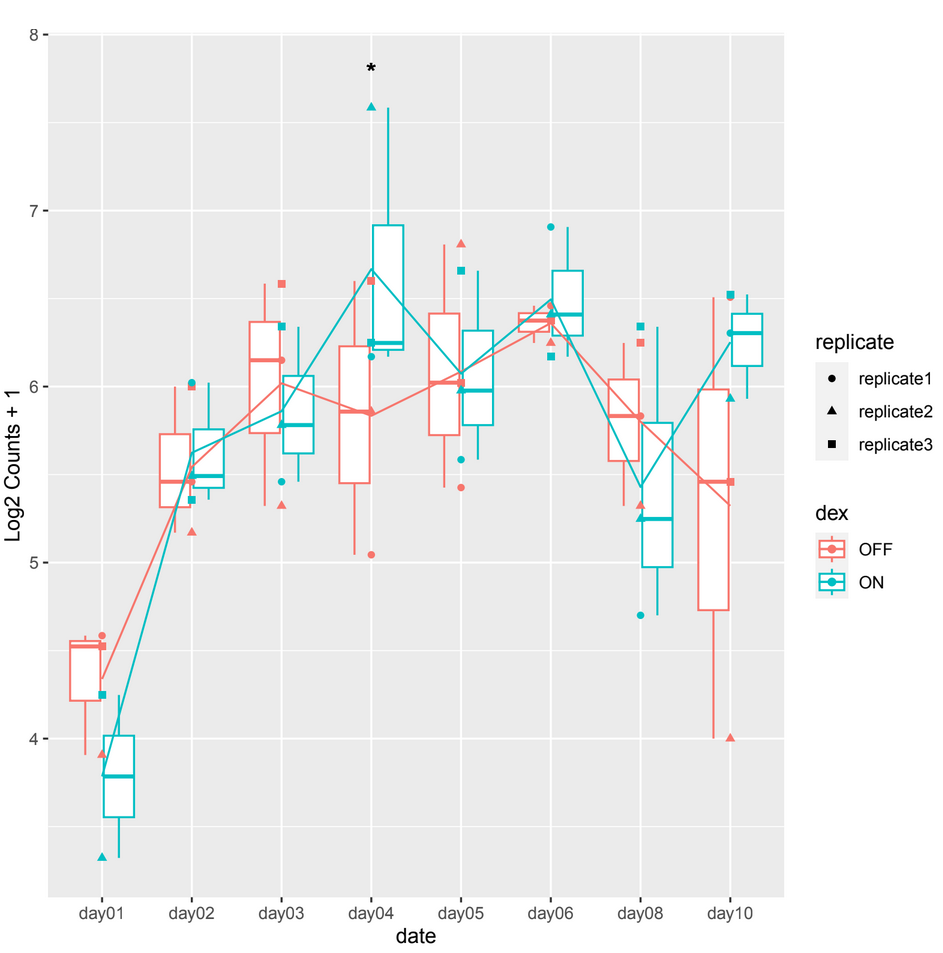
and sometimes in other count plots we observe a difference between the counts of the 2 conditions (ON/OFF) where it should be significant but it is not, such as in day10, day06 in example 2.
example 2 :
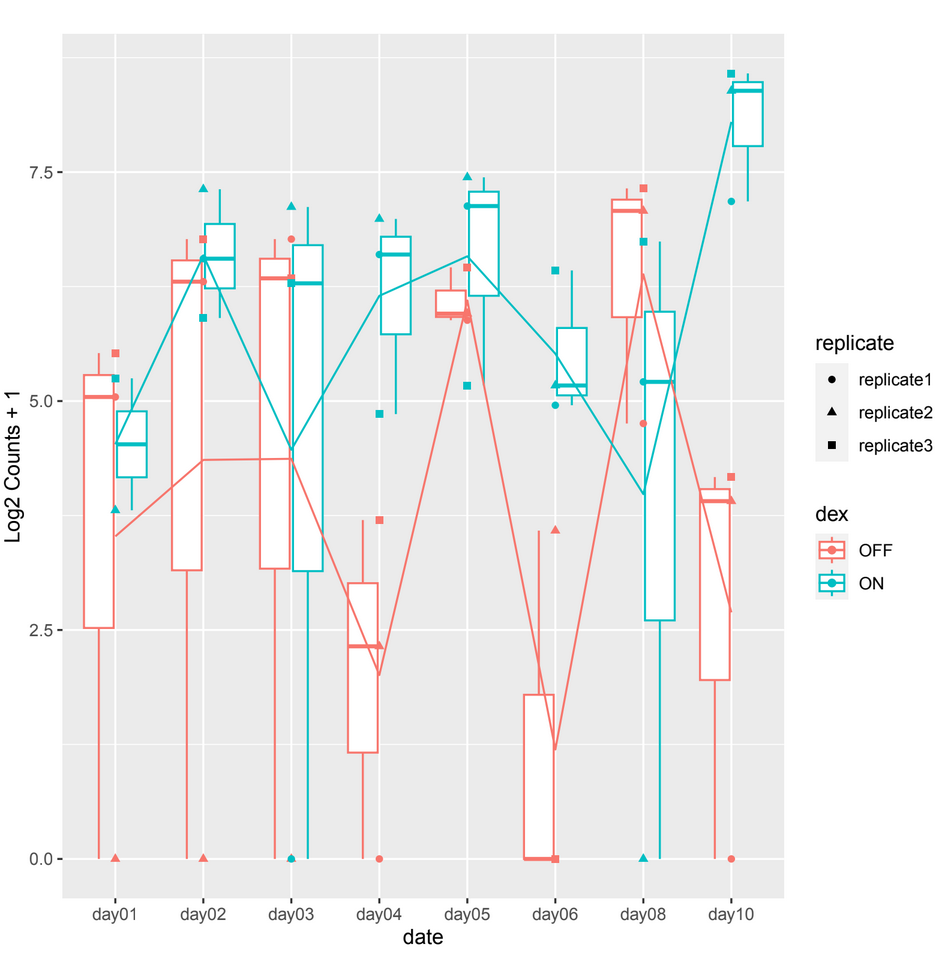
And considering the size of my dataset where significant disparities are also noticeable among the replicates, I'm concerned about potential biases or confounding factors that might be influencing these results.
Could anyone provide any insights or details regarding the potential source of the problem ?
And is there a recommended alternative approach or additional steps that I should consider when dealing with such a vast dataset in DESeq2 to mitigate potential false discoveries ?
I would greatly appreciate any insights or suggestions !

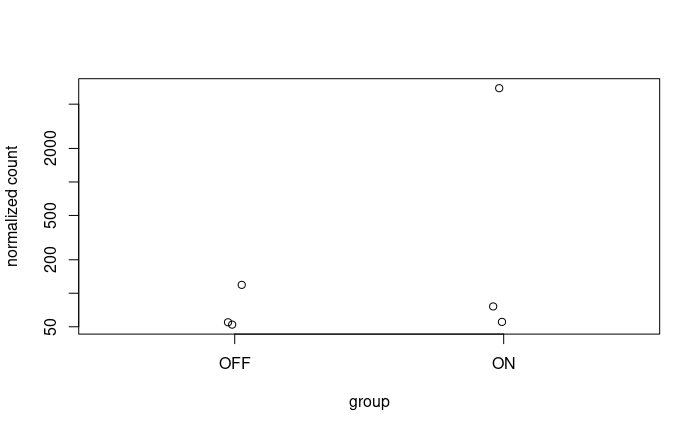
What is
Log2 Counts + 1? Please show code for the plots. And please remove the comments and move everything to the toplevel question using the Edit button. It gets messy if you spread the question like that. Right now it's the question and four comments, impossible to keep a comment structure with that.By applying the log2 transformation to the raw counts of small RNAS AND after adding 1 to each count, the resulting values often approximate a more normal distribution, which can be beneficial for downstream statistical analyses and for plotting.
and dex is the condition ON or OFF
"Raw" counts, so you're plotting un-normalized data, hence plots have no meaning in terms of groupwise comparisons. Use the
normTransformfunction or output ofvstfor plotting. That corrects for depth and composition.Thank you for your reply, but it is always the same case. Showing a significant p-adj when there isn't a difference between the 2 conditions at a certain date which is mostly on day04