
Hello, I am having an error running DESeq2 (specifically on the DESeqDataSetFromTximport step) after pseudo-aligning my transcriptomic data with Kallisto. I got the code below from this source: UT Austin Wiki
R
#load libraries
library(tximport)
library("DESeq2")
#Import a file called file_list with all the locations of the abundance.tsv files
#eg below:
#/stor/SCRATCH/sample1/abundances.tsv
#/stor/SCRATCH/sample2/abundances.tsv
#/stor/SCRATCH/sample3/abundances.tsv
#/stor/SCRATCH/sample4/abundances.tsv
files<-as.character(read.table("path/to/file_list.txt", header=FALSE)$V1)
#Import a file called samples with the sample names corresponding to each file in the file_list
#eg below:
#sample1
#sample2
#sample3
#sample4
#look at the data structures
files
#OUTPUT:
#[1] "/path/to/BMRNA2_sulcia_kallisto/abundance.tsv"
#[2] "/path/to/BMRNA4_sulcia_kallisto/abundance.tsv"
#I truncated this because I have 50 samples total
#[50] "/path/to/BMRNANo14_sulcia_kallisto/abundance.tsv"
samples<-as.character(read.table("/path/to/samples.txt",header=FALSE)$V1)
names(files)<-samples
#look at the data structures
samples
#OUTPUT:
#[1] "BMRNA2_sulcia_kallisto" "BMRNA4_sulcia_kallisto"
#truncated again...
#[49] "BMRNANo13c_sulcia_kallisto" "BMRNANo14_sulcia_kallisto"
files
#OUTPUT:
#BMRNA2_sulcia_kallisto
# "/Users/mckinleesalazar/Desktop/Dis_stuff/Data/sulcia_transcript/sulcia/just_tsv/BMRNA2_sulcia_kallisto/abundance.tsv"
#Import a file called sampletable which is a tab-delimited file that contains each samplename along with the condition
#eg below:
#samples condition
#sample1 alc
#sample2 alc
#sample3 con
#sample4 con
sampleTable <-read.table("/path/to/sampletable.txt",header=TRUE, row.names=1)
#look at the data structure
head(sampleTable)
#OUTPUT:
#Condition
#BMRNA2_sulcia_kallisto Control1
#BMRNA4_sulcia_kallisto Control2
#BMRNA12_sulcia_kallisto Control3
#BMRNA13_sulcia_kallisto Control4
#BMRNA14_sulcia_kallisto Control5
#BMRNA20_sulcia_kallisto Control6
#IMPORTANT: MAKE SURE THE SAMPLES AND FILE_LIST ARE IN THE SAME ORDER- SAMPLES SHOULD MATCH UP WITH FILES
samples==rownames(sampleTable) #should return TRUE for all
#OUTPUT:
#[1] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
#[19] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
#[37] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
#Import a file called tx2gene.csv which a csv file that contains the transcript id to gene id mapping
#For Drosophila, this is located at: tx2gene.csv
tx2gene <- read.csv("/path/to/tx2gene.csv")
#Sample of what the .csv format
#TXNAME,GENEID
#SULC_00001,50S ribosomal protein L35
#SULC_00002,Chaperone protein DnaK
#SULC_00003,50S ribosomal protein L13
#SULC_00004,30S ribosomal protein S9
#SULC_00005,30S ribosomal protein S2
#SULC_00006,tRNA-Leu(taa)
#SULC_00007,tRNA-Gly(gcc)
#look at this data structure
#read in kallisto abundance files, summarizing by gene
txi <- tximport(files, type = "kallisto", tx2gene = tx2gene)
names(txi)
#OUTPUT:
#[1] "abundance" "counts" "length" "countsFromAbundance"
#make a deseq2 object from the kallisto summarized counts
ddsMatrix <- DESeqDataSetFromTximport(countData = txi, colData = sampleTable, design = ~Condition)
ddsMatrix
#OUTPUT ERROR MESSAGE:
#Error in is(txi, "list") : argument "txi" is missing, with no default
I am not very experienced in R, but is the issue with the txi data table? I have included a screenshot of what it looks like when I view it in R studios. Is this the correct format? I.e., Should all the counts data be in a single column/row like it appears in the viewer? This doesn't seem right to be given the line:
DESeqDataSetFromTximport(countData = txi, colData = sampleTable, design = ~Condition)
It looks like it is finding the column data from 'sampleTable' but is not able to find the count data from txi. This is why I think it is a format issues in 'txi', but I cannot find an example of the expected format for the txi table.
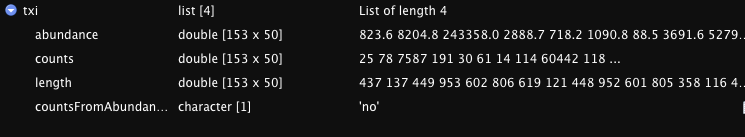
I would appreciate any help/guidance from an experienced R user. Thanks, McKinlee

