
Hi,
I'm very new to RNA seq data analysis (so almost definitely doing something stupid!). I did an R course on DataCamp on Seq analysis, where I got some working code. I have a small RNA sequencing counts data set of mature miRNA counts (un-normalised). I have made the raw counts into a matrix with the row names and column names as the genes and sample name ( they definitely are not part of the matrix). I then have a data frame of the metadata for the samples.
this is my code up to the error message:
# download DESeq
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DESeq2")
# Load library for DESeq2
library(DESeq2)
# Load library for RColorBrewer
library(RColorBrewer)
# Load library for pheatmap
library(pheatmap)
# Load library for tidyverse
library(tidyverse)
# read in data
miRNA_counts <- read.csv("mature_counts.csv")
# Explore the first six observations
head(miRNA_counts)
# Explore the structure of
str(miRNA_counts)
# transpose data frame
miRNA_counts <- t(miRNA_counts)
colnames(miRNA_counts) <- miRNA_counts[1, ]
miRNA_counts <- miRNA_counts[-1,]
head(miRNA_counts)
# Create genotype vector
Volume <- c("200", "100", "100", "200", "100", "200", "100")
# Create condition vector
condition <- c("Healthy", "Healthy", "Inflammatory", "Inflammatory", "Inflammatory", "Healthy", "Healthy")
# Create data frame
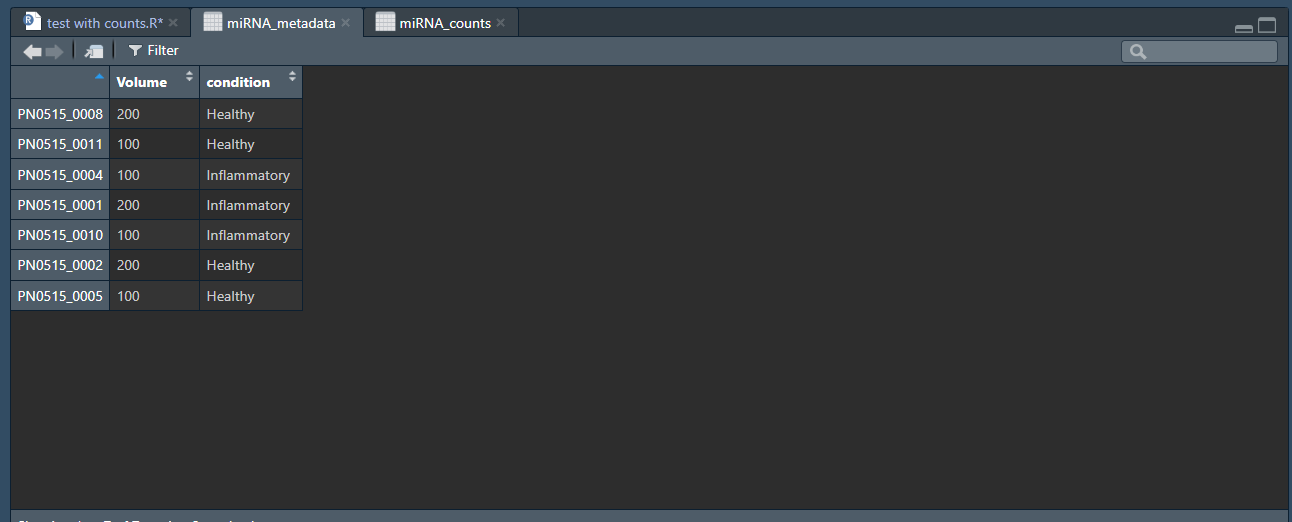
miRNA_metadata <- data.frame(Volume , condition)
# Assign the row names of the data frame
rownames(miRNA_metadata) <- c("PN0515_0008", "PN0515_0011", "PN0515_0004", "PN0515_0001", "PN0515_0010", "PN0515_0002", "PN0515_0005")
# check metadata and counts are in the same order
all (rownames(miRNA_metadata) == colnames(miRNA_counts))
# Create a DESeq2 object
dds_miRNA <- DESeqDataSetFromMatrix(countData = miRNA_counts ,
colData = miRNA_metadata,
design = ~ condition)
It is all fine up to the call of DESeqDataSetFromMatrix, where it throws an error message of "Error in DESeqDataSet(se, design = design, ignoreRank) : some values in assay are negative"
I have looked thorugh the entire dataset and there are no negative values anywhere. There are however, what i would consider weird spacing for the "0" values:
chr [1:947, 1:7] " 0" " 0" " 0" " 0" " 0" " 0" " 0" " 0" " 0" " 0" " 0" " 0" "0" ...
- attr(*, "dimnames")=List of 2 ..$ : chr [1:947] "hsa.let.7a.5p" "hsa.let.7a.3p" "hsa.let.7b.5p" "hsa.let.7b.3p" ... ..$ : chr [1:7] "PN0515_0008" "PN0515_0011" "PN0515_0004" "PN0515_0001" ...
what am I doing wrong here? I cant work it out!
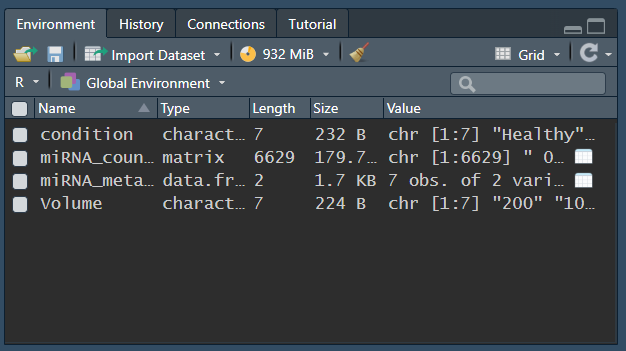
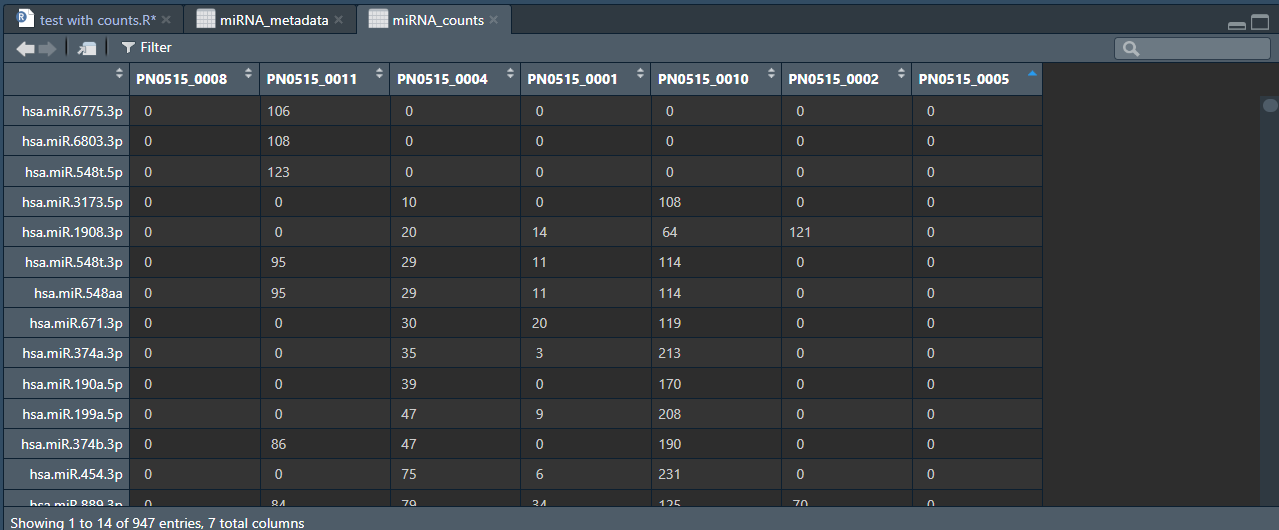

Here are some of the values for the returned call of:
I dont really understand whats going on here to be honsest, doesnt seem to really have a rhyme or reason to it throwing out a TRUE or a FALSE, unless maybe its any count number under 10 its counting as a negative??
Also when I do the code:
it returns with the same error as before "Error in DESeqDataSet(se, design = design, ignoreRank) : some values in assay are negative"
The only things I can think of is that its reading any rows with a sum of less than 10 that its thinking these are negative somehow.
I THINK I MIGHT HAVE WORKED IT OUT
for some reason the data file was read in as characters and not integers, reloaded it in with them definitely as integers and it seems to have worked (it does have a warning:
converting counts to integer mode Warning message: In DESeqDataSet(se, design = design, ignoreRank) : some variables in design formula are characters, converting to factors
but it might be good to continue on!