
I am trying to relate a factor (sensitivity) to gene expression. I have ~40 samples of breast cancer, each a different cell line, from a few lung cancer subtypes.
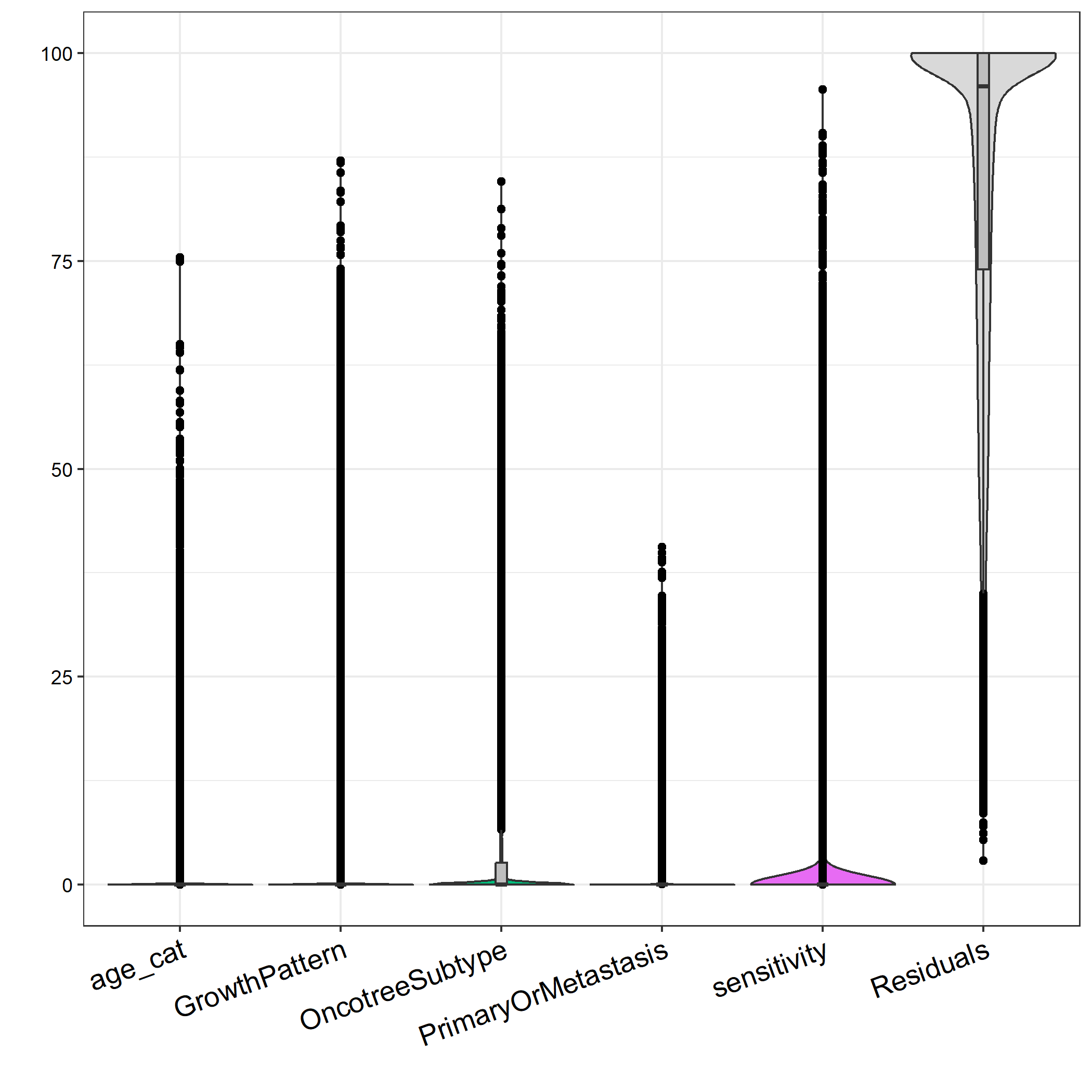
When I model my known clinical factors by variance partition to examine the variation explained from each, I see that the vast majority of my variance in gene expression is explained by unknown factors and represented by residuals. It seems my data is very noisy (which is probably to be expected as each sample comes from a different primary tumour).
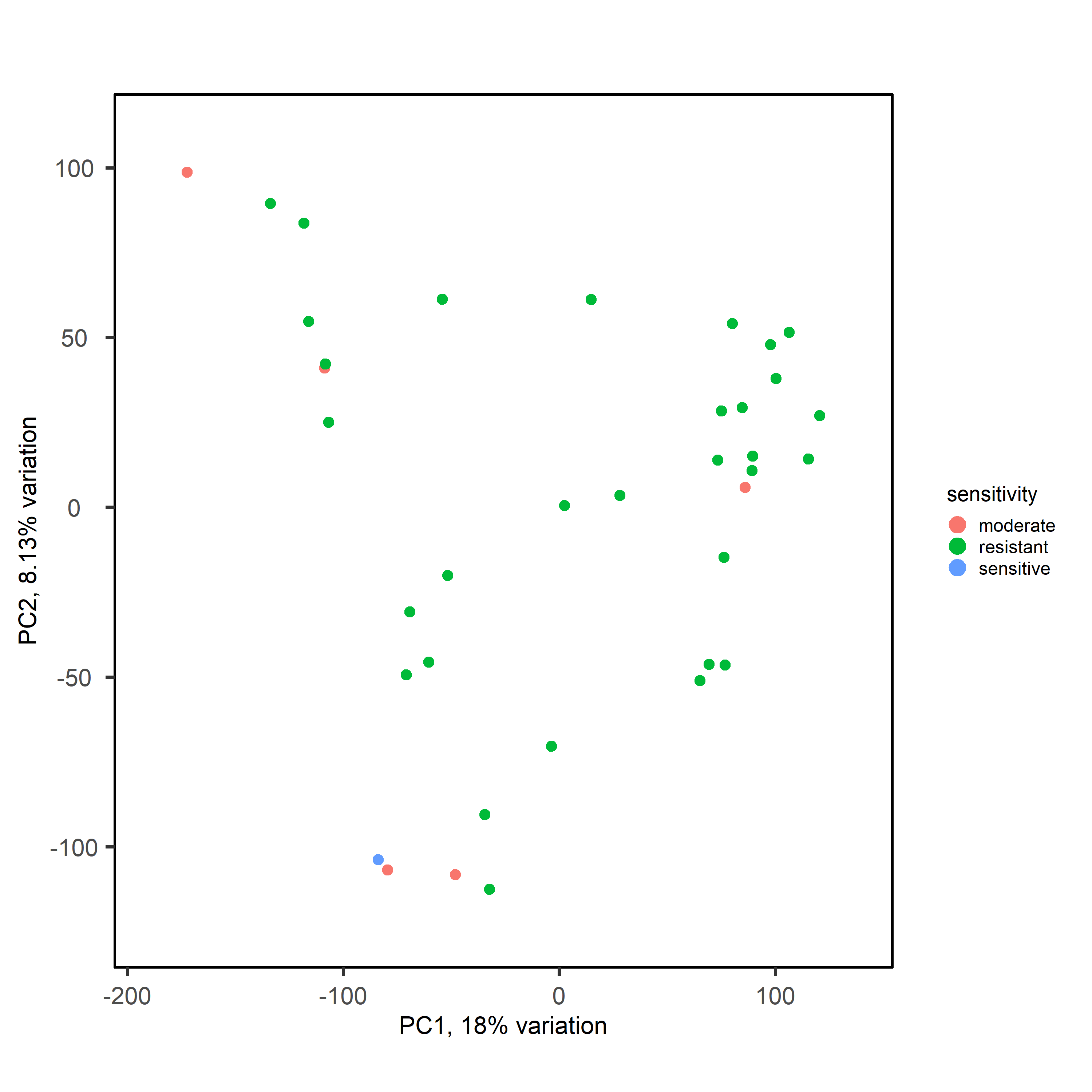
My question is: should I model these residuals / noise as unwanted variation using RUV or similar? With the aim of increasing variance explained by sensitivity, separating them strongly on PCA by sensitivity, then running DESeq with these in my design formula?
Or do I have to accept that my data is too noisy for DESeq analysis and look elsewhere for markers of sensitivity?
Crosspost on Biostars: https://www.biostars.org/p/9579592/

Amazing, thank you for your help!