
Dear Bioconductor community,
I encountered a difficult problem in correcting for batch effect in repeated measurement data, which has troubled me for a long time. This is not an experimental design usually discussed on the forum, so I'm having trouble finding solutions.
I am using cohort data, which follows the same group of healthy people and collects plasma samples every 3 years. We have performed small RNA sequencing on 1500+ plasma samples collected from multiple visits, with many of them repeated on the same individuals.
For this particular study, I want to address the following research aims:
- Assess the long-term variability of miRNA levels in human plasma
- Investigate whether the variability can be explained by different factors, as well as whether it can predict future development of health conditions (e.g., CHD) using the cohort design
As you can see, this is not a canonical group-level comparison study, but rather I intend to evaluate the within-individual between-visit variability of different miRNAs. I am thinking about using change rate (i.e., the difference in miR divided by years elapsed between 2 visits) to assess this 'variability'.
However, there seems to be some batch effect as shown in the PCA plot below:
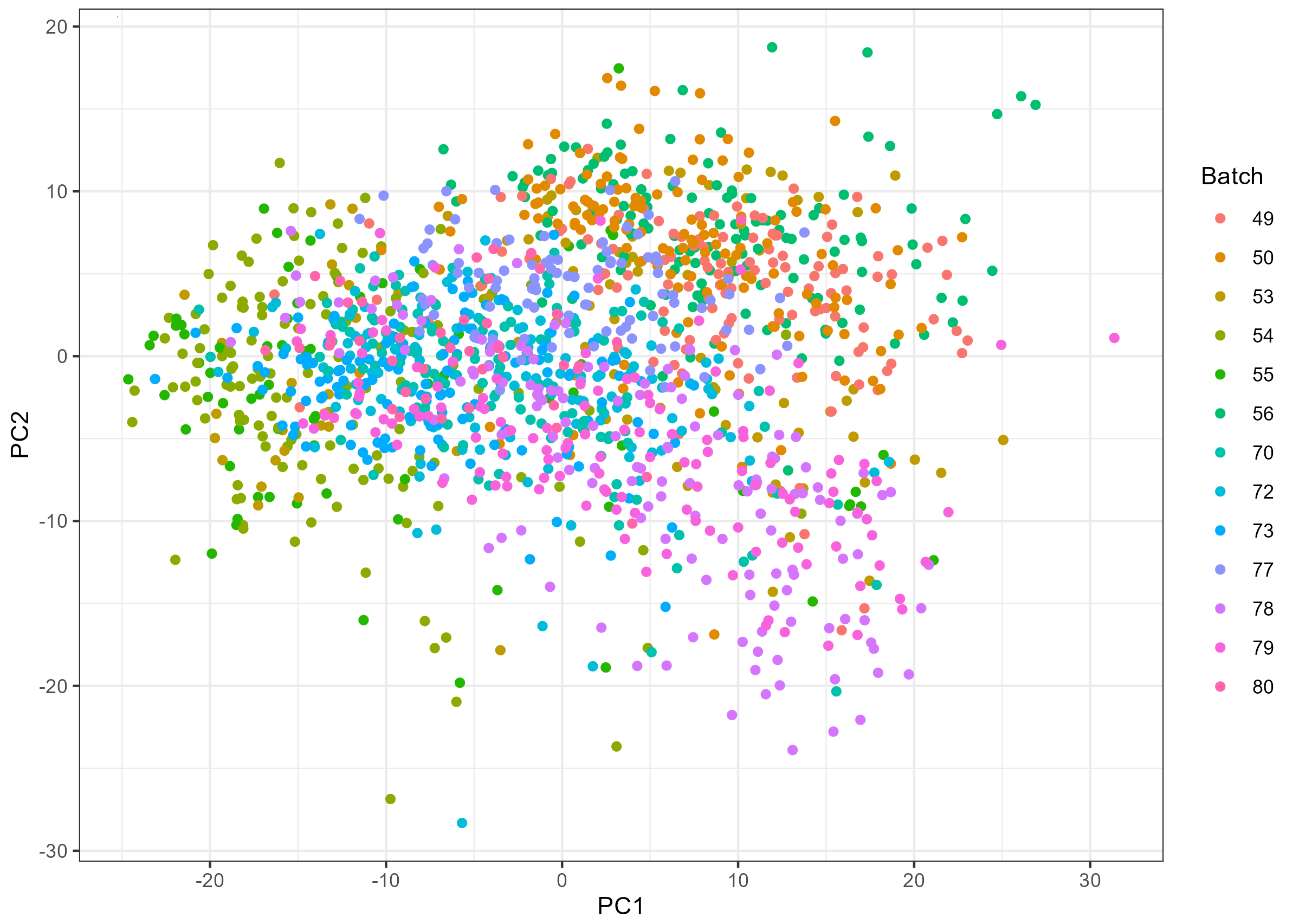
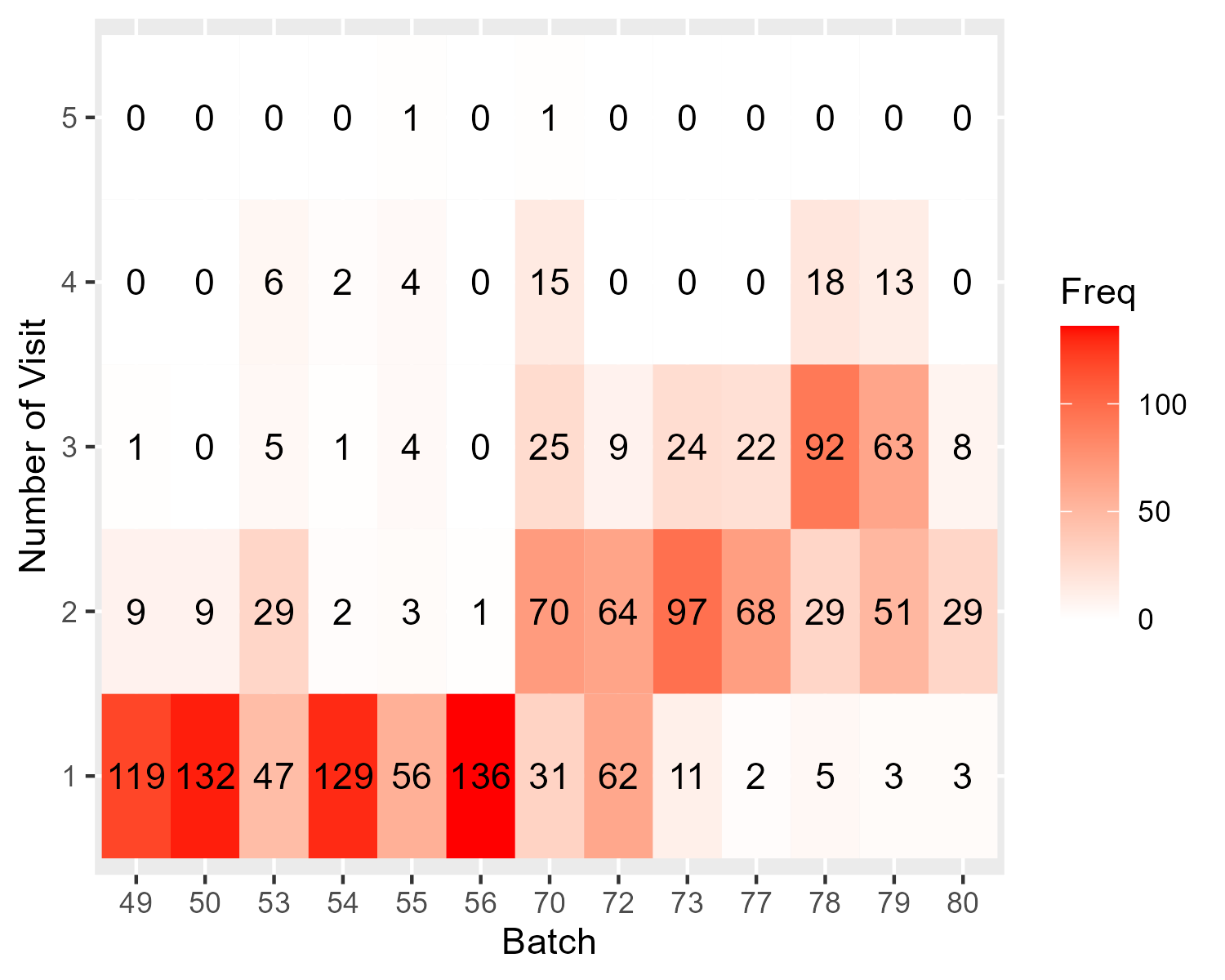
Here comes the problem: I found that samples from different visits are not evenly distributed across sequencing batches (figure shown below), so if I use batch-corrected miR data (e.g., with ComBat), I might have removed the variability between visits; if not, I might have batch effect confounded with visits.
I have also tried limma, but it seems that limma does not provide a solution to my problem, which is: how can I correct for batch effect while preserving the variability between visits (or between repeated measures)?
Thank you for your time and patience in reading this post. Any suggestions would be greatly appreciated and invaluable to me.
Best regards,
Wending

Hi James,
Thank you very much for your suggestion and inspiring comment. Unfortunately, we did not use any spike-in.
Is it possible that I adjust for the batch effect by including it as a covariate in modeling (eg, GLM model)? For example, if I calculate the difference between two visits as the response variable, can I include the combination of the two batches (as a factor variable) of the two miR measurements as a covariate in modeling?
Thank you again for your time and precious input.
Best regards, Wending
If you have two visits and one visit was measured in batch 1 and the other in batch 2, you will never be able to tell if the differences between the two visits are due to technical variability introduced by processing at two different times, or if it is due to biological variability. These things are said to be confounded.
Think of it this way. Let's say you have one person stand on a scale and you measure their weight, and then you have another person stand on a different scale, and you want to know if they weigh the same or not. The difference in their weights may be due entirely to inaccuracies in the scales (e.g., if you put the same exact weight on both and the measured weights vary), or it may be due to actual differences between the two people, or a combination of the two. Unless you weigh the two people on the same scale, or you weigh some of the people on both scales, there is no way to assess the technical differences due to the scales. You can assume that the scales are accurate and just go with the results, but if you explained to someone that you did that I am not sure they would agree with your assumption.
You have the same issue with your data. There are likely to be differences between batches, and there may be differences between subjects at the different times, but since you (or whomever) have confounded batch and subject, there is no way to determine how much of the differences are due to technical variability, and how much is due to biological variability. If you had some spike-ins, you could assume that they were accurately added (as someone who has done their fair share of pipetting, I can assure you that is a tenuous assumption), and use that as a measure of technical variability that you want to remove. You could also assume that there are a set of miRNAs that don't really change, and use
RUVgto identify an empirical set of housekeeping genes. It may actually do a great job! But you will never be able to say for sure if it did or not, because (again), your batch and subject are confounded. There is no 'one easy trick' to fix the situation, unfortunately.Thank you so much for your detailed explanation! I think I'll have to take the risk of false pos/neg given this unfortunate situation.
I am particularly grateful for your advice on using housekeeping miRNAs to adjust for batch effect. This is such wonderful advice! I will try the RUVg function. Thanks a lot!
In addition, given the ample data we have, I think we might be able to use a subset of the whole data for which the repeated measures are conducted within the same batches. Would that be a viable solution in your opinion?
Thank you again for your kind help and advice. They are truly helpful to me.
Sincerely,
Wending