Hi everyone,
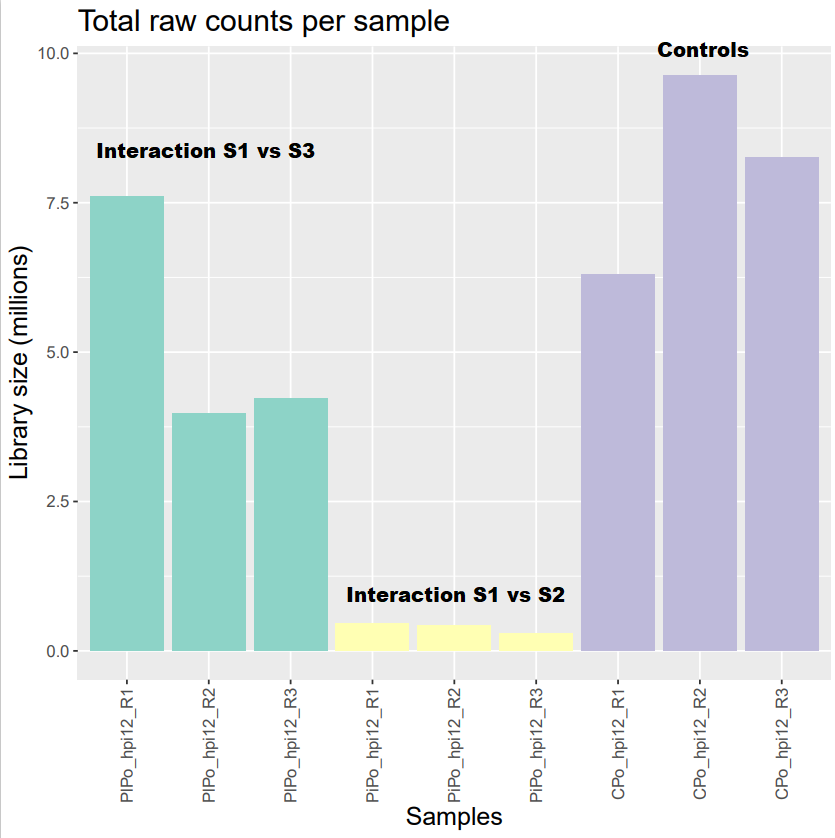
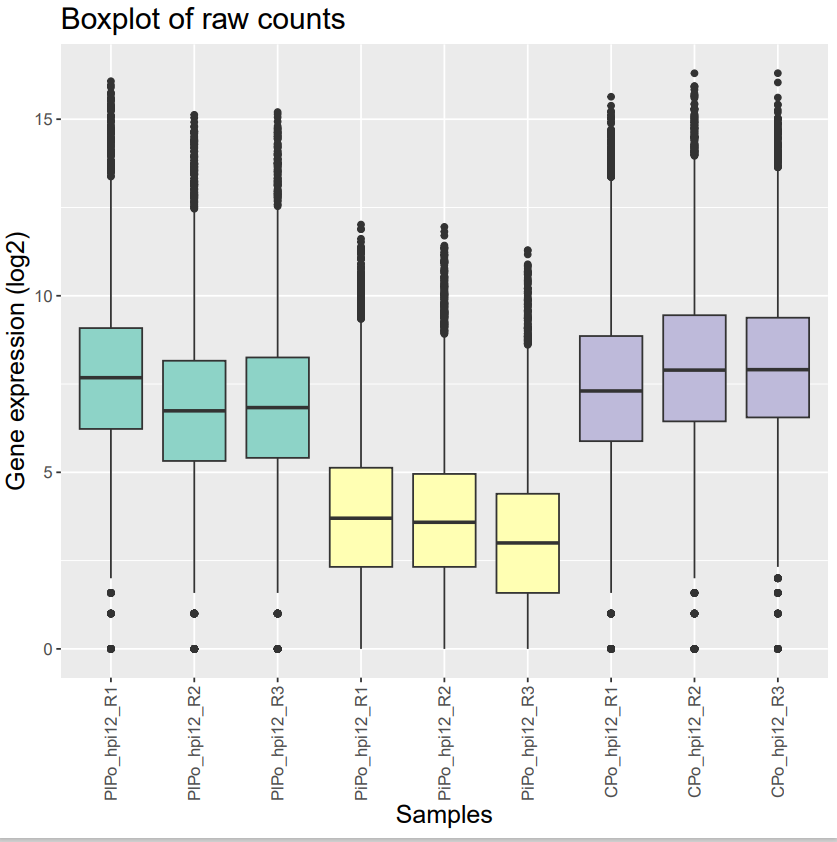
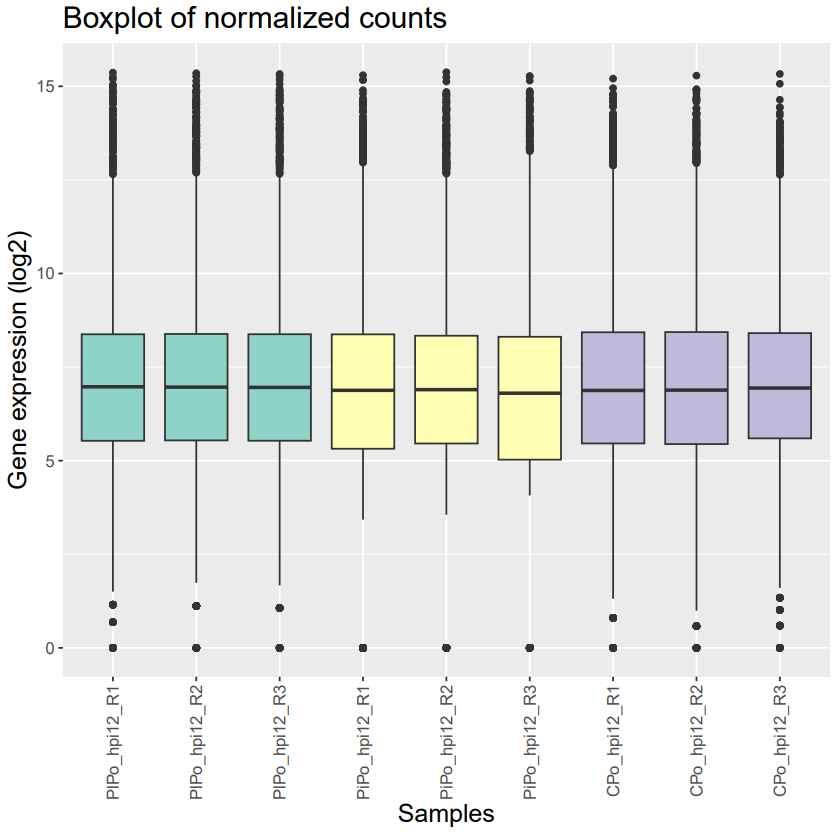
I'm working on dual species RNA-seq samples. I'm looking at 2 species (S1 vs S2 and S1 vs S3) interacting with each other and I want to compare the gene expression of S1 interacting with S2 or S3 to the gene expression of S1 growing by itself (Controls). So of course, as expected, I have huge differences of library size between the interacting samples and the control ones, especially in the condition S1 vs S2. Now I would like to know if it's possible to normalize such a huge difference in library size. I filtered my genes using a 2 CPM cut off in at least 3 samples and I normalized using both DESeq2 and EdgeR (which gave mostly the same results) and here are the results. The normalization seemed to have worked but I'm not sure if a bias was introduced and if the results that will be output after DE analysis will be reliable. Do you know if there are any methods that can deal with that kind of data ?
Thanks for your insights,
Florian