
Hi, I have dataset with transcripts from 3 population in 3 treatments and 2 replicates, totalling 18 samples.
The aim of this analysis are:
- Identify differentially expressed genes (DEGs) between pairs of treatments for each population
- Identify DEGs between populations
DESeq model ~ population + treatments + population : treatments
PCA
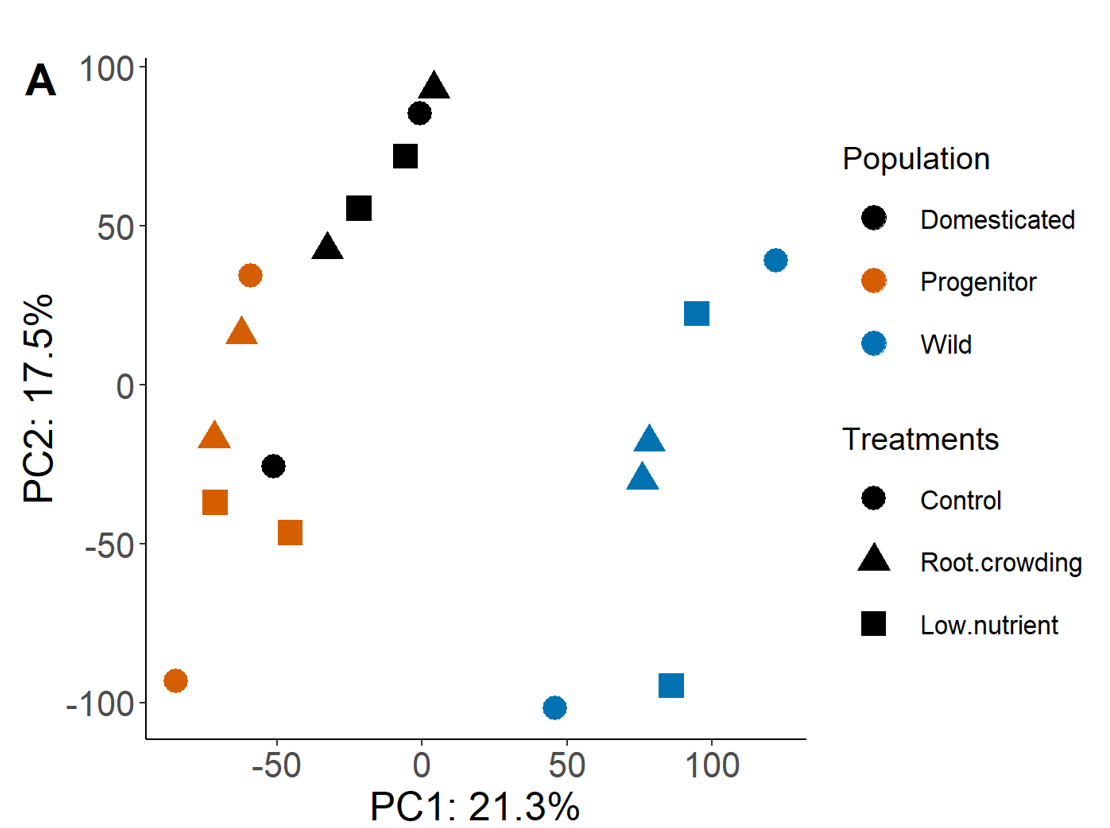
The variation within population in similar in all three, however some treatments cluster for one species but does not in another i.e. the two crowding and the two low nutrient samples cluster well in the progenitor and only one of these clusters in domesticated and wild. This clustering in the progenitor would make it easier to pick up differentially expressed genes between pairs of treatments.
I'm just wondering if it is possible to standardise the data so that there is the same variation between treatments for each population, to make identifying DEGs between populations more comparable.
Hope someone can help :) Thanks

Hi Michael, thank you for your reply.
I have tried this and the DEGs found between treatments increased for all three pairs of treatments for Domesticated and Progenitor but only increased for one pair of treatment and decreased for the other two pairs of treatment Wild.
Does this mean that subseting by population increase power for Domesticated and Progenitor analysis and decrease it for the Wild? Woud it be better to keep the analysis together?
Right, this change is expected, again from the FAQ:
https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#if-i-have-multiple-groups-should-i-run-all-together-or-split-into-pairs-of-groups
Given the variability in variance, I would do pairwise analysis. But it is up to you.
Thank you for your help! :)