Entering edit mode

Hi all,
I have a questions about bulk ATAc seq that could not find out myself and also on Biostar. I use Diffbind to get the table below (differential accessibility regions) and then use ChipPeakAnno to annotate the peaks in the P column. Could I conclude that gene with positive Fold (D columns) is more open in diseased than in control (B column vs C column)? Base on the peaks on IGV, could I say this gene in diseased is more chromatin open than the control?Thank you so much!
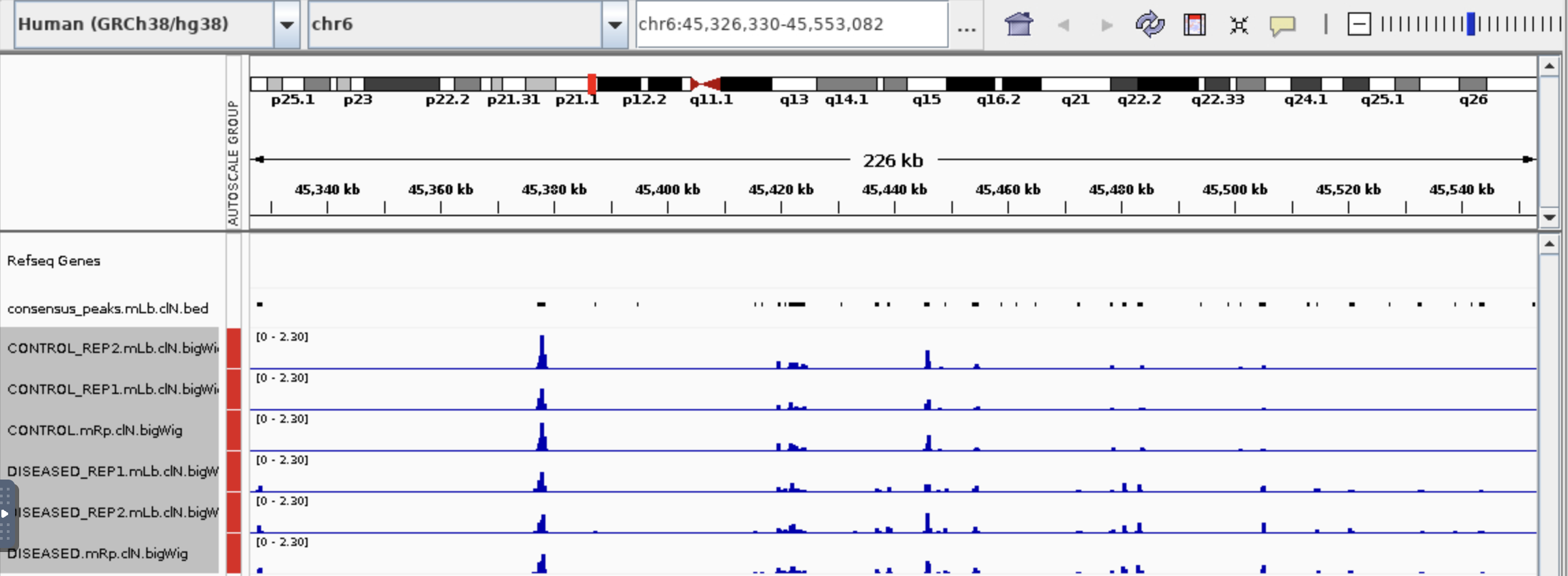


Thanks Jame for your help! It is a serious question. Sorry for chosen a bad example so I post a new one. Someone gave me advise that I need to check the value in M column to say if the gene is in open chromatin region.
Well, yes. Ideally you want the open chromatin to be near the TFBS for the gene. Open chromatin that is quite far from any known gene might be difficult to interpret.
So with distance to feature 24 in this case, could I say this gene is more open in diseased than in control? Not sure shortest distance mean, so what is the good distance to safely said a gene is in open chromatin region?
If feature 24 is the single row that is now shown, the answer is yes. But I think you already know that, as I already explained it earlier. As far as a 'good distance', that's a biological question and depends on where the TFBS sites for that particular gene are located.
Thank you for the confirmation! I am still learning ATAC seq. Chatting is not the best way but better than nothing. So if distance to feature like more than +1000 or less than -1000, I could not conclude if the genes are in open chromatin region, is that correct?