afer reading some posts about different DE analysis provided by edgeR, I found that the QL framework it's a the better choice, as it provides more accurate type I error rate control.
However, when the dispersions are very large and the counts are very small it's better to switch to the LRT framework.
But what do you mean for "very large" and "very small"? Are there reasonable thresholds?
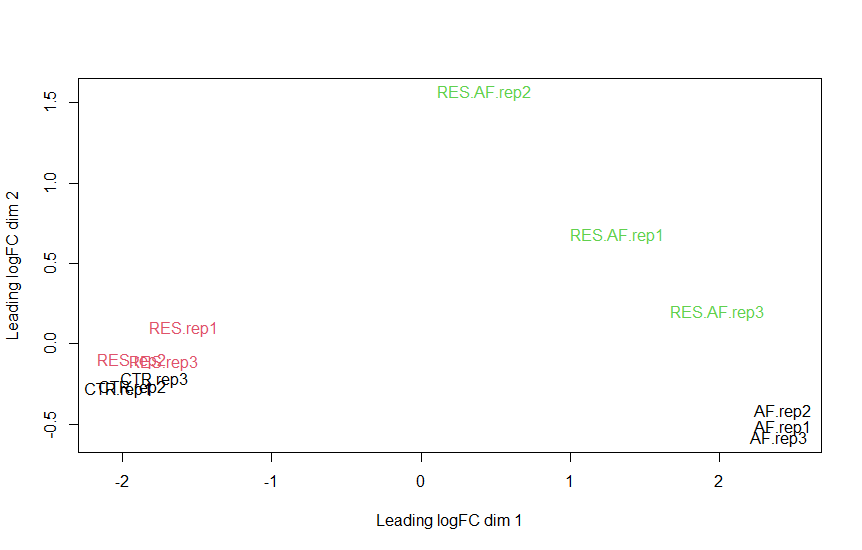
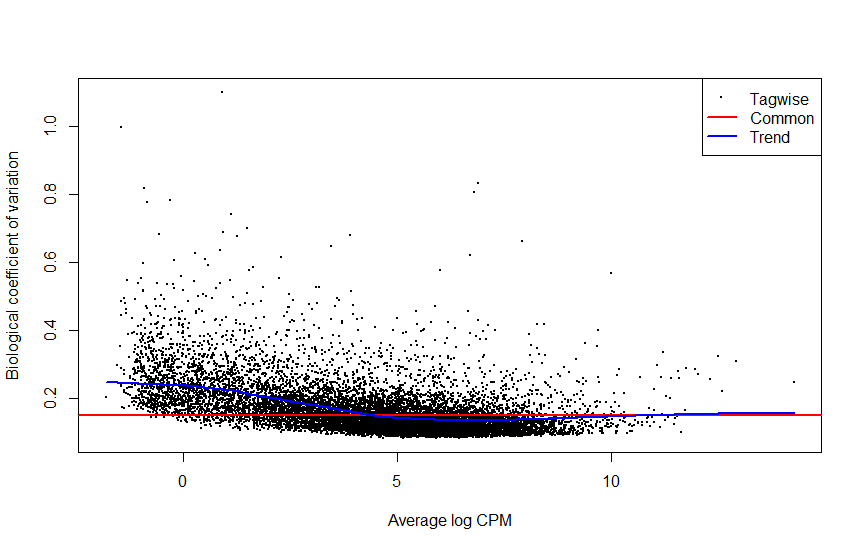
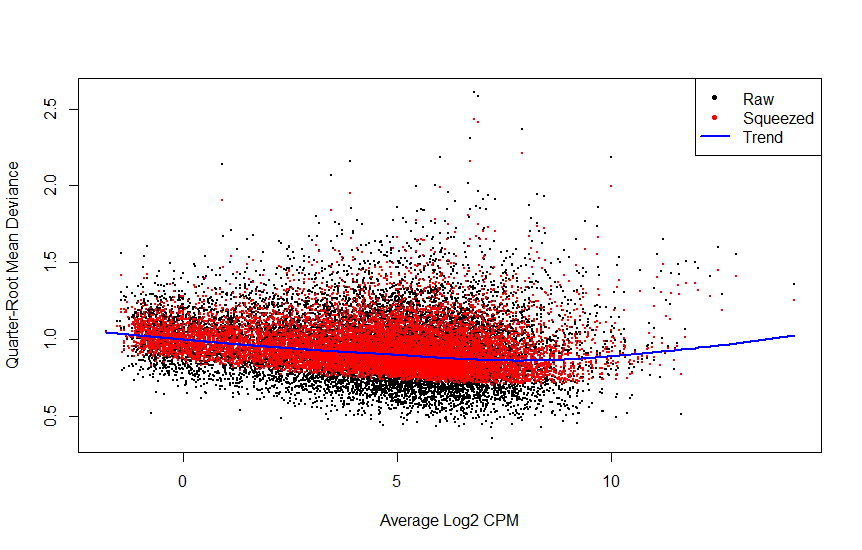
For instance, I'm dealing with a dataset of 12 samples (1 variable/4 factors), and I'm not sure about the best approach to be adopted.
Based on plotMDS, plotBVC, and plotQLDisp posted above, what's your feeling?
Thank you in advance for your time and suggestions!
Best
Marianna
In my opinion, you should simply be using the quasi-likelihood pipeline. For a regular bulk RNA-seq experiment with 12 samples you should virtually always be using glmQLFit and glmQLFTest.
when the dispersions are very large and the counts are very small it's better to switch to the LRT framework.
The edgeR documentation does not recommend this. You seem to be quoting Aaron Lun from here ( EDGE-R exact test vs QL F-test ). I will let Aaron Lun respond -- he might have had a single cell or ChIP-seq context in mind.
he might have had a single cell or ChIP-seq context in mind.
Yes, single-cell. I was fiddling with Smart-seq2 read count data - dispersions routinely greater than 1, average counts less than 1. IIRC the QL tests were not holding their size on simulated data; I think I traced the cause to a deterioration in the saddlepoint approximation such that the deviances were not chi-squared distributed. A toy demonstration is provided below:
library(edgeR)
y <- matrix(rnbinom(5000000, mu=0.5, size=0.5), ncol=50)
y <- DGEList(y)
design <- model.matrix(~gl(2, 25))
fit <- glmQLFit(y, design, dispersion=2) # using the true NB dispersion.
res <- glmQLFTest(fit, coef=2)
mean(res$table$PValue <= 0.01)
## [1] 0.03035
mean(res$table$PValue <= 0.05)
## [1] 0.09861
The issue disappears at larger means and/or smaller dispersions, so is largely irrelevant to discussions of bulk RNA-seq data analysis.
At the time, I ended up using the LRT, but further contemplation led me to realize that there were more fundamental problems with treating cells as replicates of each other. Hence the use of pseudo-bulk samples, which (i) captures the right level of replication without the need to use complex mixed models and (ii) increases the counts and decreases the dispersions such that QL methods can be safely applied (e.g., scran::pseudoBulkDGE).
For ChIP-seq, the QL method can be used without much concern. The counts are usually high enough for regions of interest (usually enriched peaks) and the dispersions in the same ballpark as bulk RNA-seq.
For the benefit of other readers, let me clarify that all the data rows in the above simulation would be filtered out by filterByExpr and so would not appear in a bulk RNA-seq analysis.
It seems for bulk RNA-seq analysis the QL approach should be always preferred over LRT.
Thus, based also on the manual, likelihood ratio test is an option in single cell RNA-seq analysis and when no replicates are available.
Hi there,
I am "resurrecting" this post cause I have found it while searching for an explanation to understand my RNAseq results.
I was using the glmFit followed by glmLRT but, as advised by Gordon here, I switched to the QL pipeline (by running glmQLFit followed by glmQLFTest).
I've got a similar situation that @Marianna had: I have a dataset of 16 samples (1 variable/4 factors/ 4 replicates per factor). I have tried the QL pipeline. In short I import the count table with all the samples, filter it via filterByExpr using the default values, then after having estimated the dispersion with estimateDisp and fitted the QL via glmQLFit I run pairwise comparisons via glmQLFTest. Problem is I always end up getting no differentially expressed genes. I tried several contrasts (since I have 4 factors) but it doesn't change the fact that I always have 0 diff. expressed genes no matter which contrast I test.
I should add that my dataset was generated using the Lexogen QuantSeq 3' tag sequencing technology and I had about 5 million raw reads per sample (give or take).
By reading the replies to this post I understood that the QL pipeline is not well suited for cases where the counts are small (like the example by @Aaron where the average expression per sample was below 1). I then checked my raw count table and found that the average expression level per sample is about 16. Is it possible that in my situation, the "low" average expression level is not enough for the QL pipeline?
Looking forward to hearing your feelings about it
Thanks again
Luca
You are posting an answer to a question from 2.3 years, but in reality your post is not an answer at all but rather a new question. I will answer your question this time but, in future, please open a new question rather than making a pretend-answer to an old thread. In reality, you are continuing your question from here: Question about filterByExpr function edgeR
Getting 0 DE genes is not an error. Quite the contrary, edgeR is deliberately written so that it only gives significant DE genes when there is genuine evidence of reproducible DE.
Our experience is that edgeR works very well with Lexogen QuantSeq 3' tag sequencing technology and about 5 million reads per sample is just fine. An average count of 16 is no problem at all. This is exactly the sort of data that edgeR QL is designed for.
The current version of edgeR QL has trouble with single-cell data but it is just fine for almost any sort of bulk RNA-seq. Even for single-cell data, edgeR QL tends to give too many DE genes rather than too few.
As I advised you before, you should be exploring the quality of your data. If you are not getting DE when you expect to, then you should be looking for outliers or batch effects or trouble-shooting why your data is overdispersed (many zeros mixed with a few large counts for the same gene). Blaming the DE test is not likely to help. From the data example you showed before, I suspect that edgeR QL test is very likely reflecting correctly the variability in your data.



Yes, single-cell. I was fiddling with Smart-seq2 read count data - dispersions routinely greater than 1, average counts less than 1. IIRC the QL tests were not holding their size on simulated data; I think I traced the cause to a deterioration in the saddlepoint approximation such that the deviances were not chi-squared distributed. A toy demonstration is provided below:
The issue disappears at larger means and/or smaller dispersions, so is largely irrelevant to discussions of bulk RNA-seq data analysis.
At the time, I ended up using the LRT, but further contemplation led me to realize that there were more fundamental problems with treating cells as replicates of each other. Hence the use of pseudo-bulk samples, which (i) captures the right level of replication without the need to use complex mixed models and (ii) increases the counts and decreases the dispersions such that QL methods can be safely applied (e.g.,
scran::pseudoBulkDGE).For ChIP-seq, the QL method can be used without much concern. The counts are usually high enough for regions of interest (usually enriched peaks) and the dispersions in the same ballpark as bulk RNA-seq.
For the benefit of other readers, let me clarify that all the data rows in the above simulation would be filtered out by
filterByExprand so would not appear in a bulk RNA-seq analysis.