
Hi, I'm using DESeq2 to run analysis on 3 different cell types undergoing the same treatment conditions, each with 2 replicates (see metadata below). However, 1 of the cell types (HCT116) was done in a different lab and shows a stark batch effect between its two replicates, while the other 2 cell types have a more muted batch effect between their two replicates (PCA below).
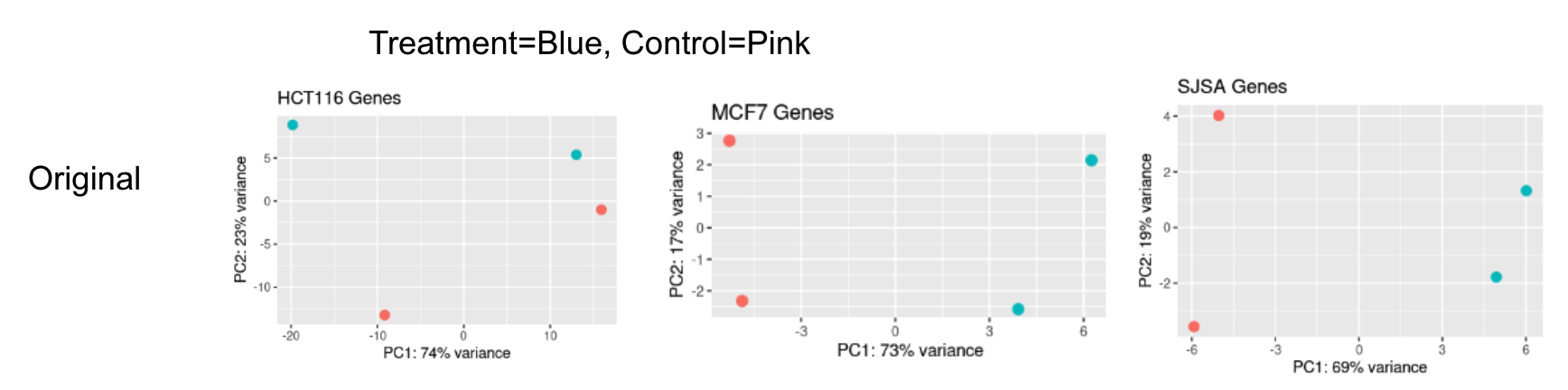
Downstream, we are most interested in identifying the DE genes that are shared and unique between cell types.
I've analyzed each cell type in their own separate DESeq2 object using both replicate+treatment and treatment designs. Overall, I see the best precision with a known set of gene targets when using the native DESeq2 batch correction in HCT116, but the other two cell types show a small loss of TPs when using the ~replicate+treatment design. To me, this suggests that I'm fitting noise with the batch term in only these two cell types, which is causing these issues.
Right now, I'm considering integrating all of the datasets into a single DESeq2 object with the metadata table above, and then identifying the shared and cell-type specific response genes from this run using the design recommended at this link: (~cell + cell:rep + cell:treatment, section "Group-specific condition effects, individuals nested within groups" of the DESeq2 vignette), and then employing the appropriate group contrasts. Is simply finding the overlap of the individual DESeq2 runs (selectively applying batch correction or just using a batch correction for all) my best option, or is grouping them together the more robust method? The output statistics from the latter could also be useful for the planned analysis downstream, but I'm worried about losing cell-type specific signal with this design, plus fitting noise due to the batch issues stated above. Which is the better approach?
Thank you and let me know if you have any follow up questions!

