
Hello! I am using DESeq2 to compare three cell types that are part of a cancer progression model. The cells share a similar genetic background, but there are karyotypic differences between the cell lines. In particular, each cell line has its own chromosome trisomies. I imagine that these trisomies would affect counts in a chromosome-specific manner, and I want to be sure I am handling them right.
I am using DESeq2 to compare Hi-C contacts at loops called among these three cell types. I have an un-normalized count matrix of Hi-C contacts from 29,770 total loops across 24 samples (8 per cell line).
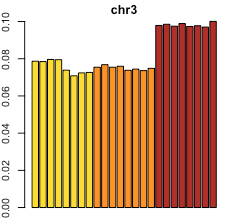
Looking at the percentage of total loop counts per chromosome, you can see evidence of the trisomies in the counts themselves; for example, this plot below shows that all 8 replicates from the most cancerous cell line [in red] have a higher percentage of counts from chromosome 3 than the other 2 cell lines do. This is consistent with the fact that this cell line has trisomy 3, while the other two [in yellow, orange] do not.
I also noticed that after running DESeq2, we have a higher number of differential loops on chromosomes that have cell type-specific trisomies. For example, chr3 has a trisomy in one cell line while chr2 and chr4 do not. On chromosomes 2, 3, and 4, we have called 2604, 2225, and 2092 loops respectively. However, we see 148, 415, and 113 differential loops respectively, with chr3 having a much larger proportion of differential loops than chr2 or chr4.
Overall, many of our differential loops look very convincing and I am happy with how DESeq2 is performing here. However I'm concerned that some of our differential loops are a result of there being more reads across an entire chromosome for one sample (or large regions of it) rather than local differences that we are more interested in. I want to be sure we are utilizing DESeq2 to its fullest extent here and capturing as many biologically significant changes as we can.
I know that DESeq2 internally corrects for library size, but I wasn't sure about how it could handle chromosome-specific differences in sequencing depth. I have been trying to think of ways to address this, but would appreciate any insight from someone who understands the software more deeply. So far my thoughts are:
- Would something like providing custom normalization factors be an appropriate way to correct for chromosome-specific differences in sequencing depth? I haven't used them before and I'm not sure where I would get them from in this case, but it seemed like the closest option I could find from the documentation.
- Would it be appropriate to run DESeq with the loops from trisomy chromosomes subset out? In turn, would it be appropriate to run DESeq on the trisomy chromosomes individually, or would reducing the size of the input matrix so much be problematic (the smallest chromosome has only 287 loops)? While it wouldn't be ideal to drop entire chromosomes from my analysis, we could work with it if that's what would make the most sense, since only 5 or so chromosomes are affected.
- Do you think it would be best to just move forward with the results I have, instead? While my understanding of the data and DESeq led me to believe this would cause issues, I would not be surprised if in reality it is less impactful than I'd think.
For reference, I have been running DESeq (version 1.38.3, R version 4.2.1) as follows:
# Create coldata
colData <- data.frame(cell = factor(rep(c("A", "T", "C"), each = 8)),
brep = factor(rep(rep(c(1, 2), each = 4), times = 3)),
trep = factor(rep(c(1:4), times = 6)))
rownames(colData) <- colnames(countMatrix)
# Create DESeq Data Set from matrix
dds <- DESeqDataSetFromMatrix(countMatrix,
colData = colData,
design = ~ trep + brep + cell)
dds <- DESeq(dds,
test = "LRT",
full = ~ trep + brep + cell,
reduced = ~ trep + brep)
I'm very sorry in advance if this is something that was already addressed in the vignette or previous questions that I just missed! Thanks for your time and any help you can provide!
-Katie

Hi Mike!
I'm sorry to be so slow to respond, but thanks so much for the help! I'm glad to know I was on the right track with the normalization factors, and it's very easy to use! This looks like it will indeed suit our needs perfectly. I just need to figure out the best way to generate that
dnacopymatrix as accurately as possible, but that is our own mess to worry about. (We have a few options, with the % of reads per chromosome data we have, some orthogonal karyotype data, and even Hi-C expected counts might be an option).I will play around with different inputs for that matrix but I'm sure one of those will help relieve the bias we're seeing! Immediately with just some estimated numbers it seems to still call a few more differential loops on those chromosomes (maybe this is in part due to their simply being deeper sequencing on those chromosomes due to the karyotypic differences?), it's less extreme of a difference. The differential loops called are also much more balanced in terms of whether they are increasing or decreasing over the system, whereas before there was a strong bias one direction or another depending on which samples had the extra chromosomes, which I think is a great sign!
Thanks again for all your help!
that certainly could be true: having more reads (even after correcting the bias issue) would give you more power to detect DE.