Entering edit mode

Hi everyone,
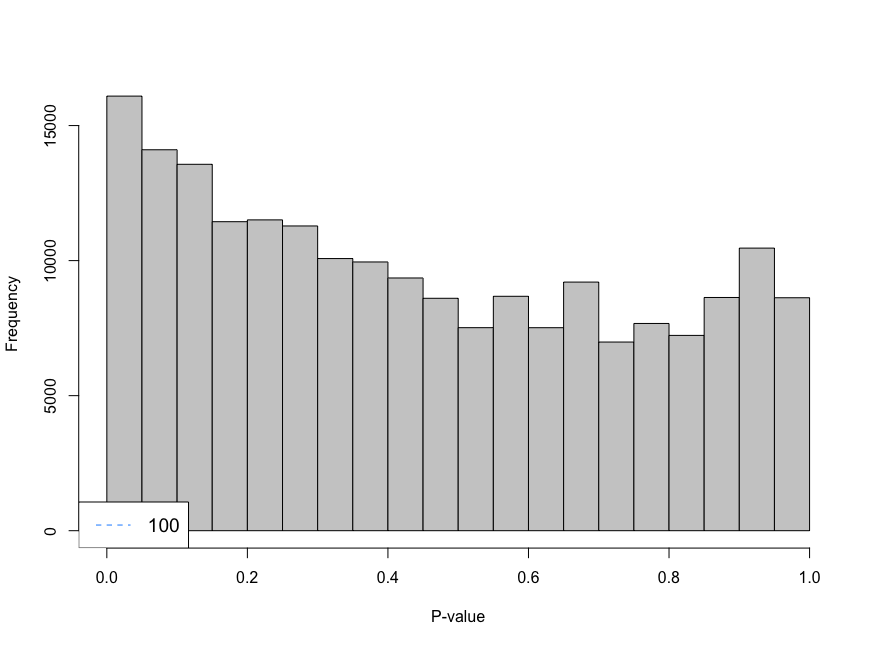
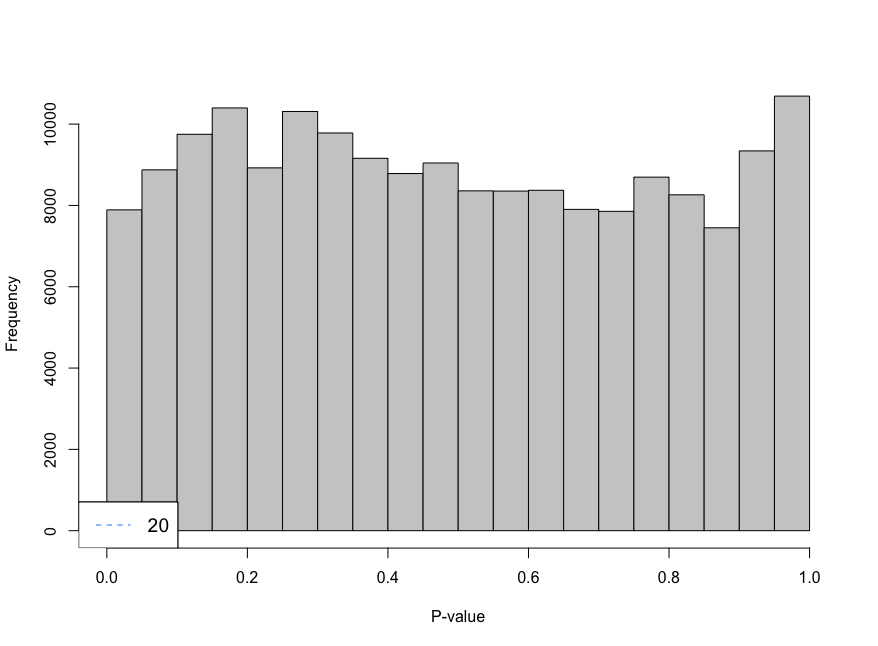
I am trying to analyse single cell data using CellRanger unfiltered data and I am using emptyDrops to remove droplets that contain ambient RNA. When using the parameter lower I am using 100 as the limit, however, the histogram shows large peaks near cero. I have decreased the parameter lower from 100 to 20 and look at the histogram. The histogram gives an "even" distribution at 20. I am not sure if that is a too low value to use as limit.
-Is decreasing the limit a sensible choice? -Not sure, if I fully understand the effect of setting a limit for this function -Is there any function to measure the lower limit?
Code should be placed in three backticks as shown below
limit <- 20
e.out_sample_1 <- emptyDrops(counts(sample_1), lower=limit, test.ambient=TRUE, BPPARAM = bp.params)
hist(e.out_sample_1$PValue[e.out_sample_1$Total <= limit & e.out_sample_1$Total > 0],
xlab="P-value", main="", col="grey80")
legend("bottomleft", legend=limit,
col="dodgerblue", lty=2, cex=1.2)
sessionInfo( )
R version 4.2.2 (2022-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.3.1
Matrix products: default
LAPACK: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] lubridate_1.9.2 forcats_1.0.0 stringr_1.5.0
[4] dplyr_1.1.2 purrr_1.0.1 readr_2.1.4
[7] tidyr_1.3.0 tibble_3.2.1 tidyverse_2.0.0
[10] UpSetR_1.4.0 cowplot_1.1.1 BiocParallel_1.32.6
[13] scater_1.26.1 ggplot2_3.4.2 scuttle_1.8.4
[16] DropletUtils_1.18.1 SingleCellExperiment_1.20.1 SummarizedExperiment_1.28.0
[19] Biobase_2.58.0 GenomicRanges_1.50.2 GenomeInfoDb_1.34.9
[22] IRanges_2.32.0 S4Vectors_0.36.2 BiocGenerics_0.44.0
[25] MatrixGenerics_1.10.0 matrixStats_0.63.0
loaded via a namespace (and not attached):
[1] bitops_1.0-7 bit64_4.0.5 httr_1.4.5
[4] tools_4.2.2 utf8_1.2.3 R6_2.5.1
[7] irlba_2.3.5.1 HDF5Array_1.26.0 vipor_0.4.5
[10] DBI_1.1.3 colorspace_2.1-0 rhdf5filters_1.10.1
[13] withr_2.5.0 tidyselect_1.2.0 gridExtra_2.3
[16] bit_4.0.5 compiler_4.2.2 cli_3.6.1
[19] BiocNeighbors_1.16.0 DelayedArray_0.24.0 scales_1.2.1
[22] R.utils_2.12.2 XVector_0.38.0 pkgconfig_2.0.3
[25] sparseMatrixStats_1.10.0 fastmap_1.1.1 limma_3.54.2
[28] rlang_1.1.0 rstudioapi_0.14 RSQLite_2.3.1
[31] DelayedMatrixStats_1.20.0 generics_0.1.3 R.oo_1.25.0
[34] RCurl_1.98-1.12 magrittr_2.0.3 BiocSingular_1.14.0
[37] GenomeInfoDbData_1.2.9 Matrix_1.5-4 Rcpp_1.0.10
[40] ggbeeswarm_0.7.1 munsell_0.5.0 Rhdf5lib_1.20.0
[43] fansi_1.0.4 viridis_0.6.2 lifecycle_1.0.3
[46] R.methodsS3_1.8.2 stringi_1.7.12 edgeR_3.40.2
[49] zlibbioc_1.44.0 rhdf5_2.42.1 plyr_1.8.8
[52] blob_1.2.4 parallel_4.2.2 ggrepel_0.9.3
[55] dqrng_0.3.0 crayon_1.5.2 lattice_0.21-8
[58] Biostrings_2.66.0 beachmat_2.14.2 hms_1.1.3
[61] KEGGREST_1.38.0 locfit_1.5-9.7 metapod_1.6.0
[64] pillar_1.9.0 igraph_1.4.2 codetools_0.2-19
[67] ScaledMatrix_1.6.0 glue_1.6.2 scran_1.26.2
[70] remotes_2.4.2 BiocManager_1.30.20 png_0.1-8
[73] vctrs_0.6.2 tzdb_0.3.0 gtable_0.3.3
[76] cachem_1.0.7 rsvd_1.0.5 viridisLite_0.4.1
[79] AnnotationDbi_1.60.2 beeswarm_0.4.0 memoise_2.0.1
[82] cluster_2.1.4 statmod_1.5.0 bluster_1.8.0
[85] timechange_0.2.0
Thanks!!
