Entering edit mode

I have a complex design where I have 3 karyotypes. Each genotype has samples from different patients (patient_name in the metadata), and from each patient they derived different clones. I am interested in the effect of the karyotype (47vs46, and 49vs46), taking into account the patient origin and clone origin.
This is my metadata:
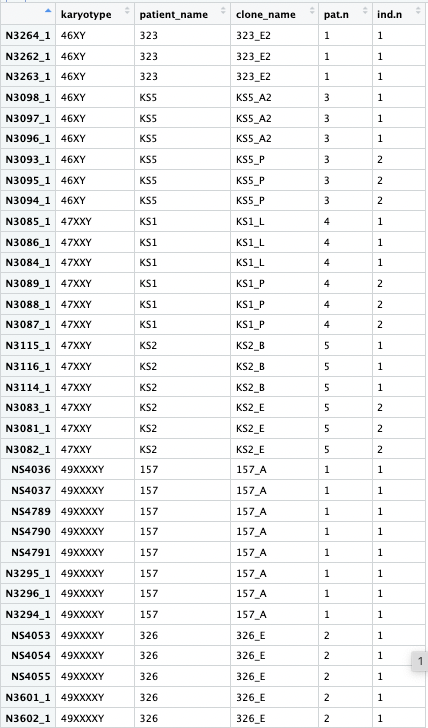
I try to create my model with a custom matrix by doing like this:
mod.matrix=model.matrix(~ karyotype:pat.n:ind.n + karyotype, metadata_NSC)
mod.matrix=mod.matrix[,which(colMeans(mod.matrix)!=0)]
dds_NSC <- DESeq(dds_NSC, betaPrior = FALSE, full=mod.matrix)
However I get a not full rank model error:
Error in checkFullRank(full) :
the model matrix is not full rank, so the model cannot be fit as specified.
One or more variables or interaction terms in the design formula are linear
combinations of the others and must be removed.
Am I using the nesting wrongly?
PS. The clones are unfrozen independently and therefore are not proper technical replicates (even if they derive from the same patient) Thanks
