Entering edit mode

Hi,
I am trying to create a txi list with transcript data from running Salmon. I have created a tx2gene file with 2 columns with protein product and locus tag taken from NIH. However i am getting the following error when running the command for tximport
txi <- tximport(files, type="salmon", tx2gene=tx2gene)
error:
reading in files with read_tsv
1 Error in tximport(files, type = "salmon", tx2gene = tx2gene) :
all(c(abundanceCol, countsCol, lengthCol) %in% names(raw)) is not TRUE
Any help would be appreciated. Thanks JD

Thank you for getting back to me. I am new at RNAseq analysis so not sure if i am doing it the right way.
In order to do get the transcriptome file to run salmon i took the fasta file with coding sequences from this NIH https://www.ncbi.nlm.nih.gov/nuccore/197361212?report=fasta and attached an image of the file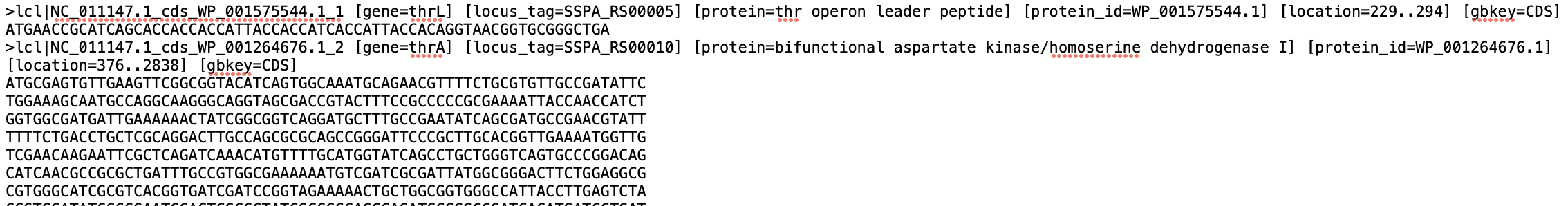 and to get the list of transcript -> gene i got the 2 columns of Protein Product (transcript) and Locus tag (gene id) from here https://www.ncbi.nlm.nih.gov/genome/browse/#!/proteins/152/299238%7CSalmonella%20enterica%20subsp.%20enterica%20serovar%20Paratyphi%20A%20str.%20AKU_12601/ and an image form R is here
and to get the list of transcript -> gene i got the 2 columns of Protein Product (transcript) and Locus tag (gene id) from here https://www.ncbi.nlm.nih.gov/genome/browse/#!/proteins/152/299238%7CSalmonella%20enterica%20subsp.%20enterica%20serovar%20Paratyphi%20A%20str.%20AKU_12601/ and an image form R is here 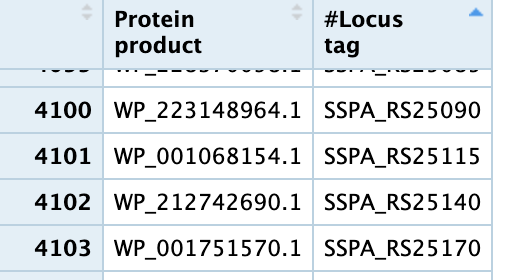 .
.
The only thing i changed in the quant.sf files was the name of the transcripts (first column) as they came out as the full name from transcriptome file - everything after '>' and so i just shorten them to match the transcript name column of tx2gene file.
Could you please tell me if you can tell where i went wrong on this?
Thank you once more
tximport reads in the files as output by Salmon. If you manually changed the files and their headers you will have to tell tximport your new columns names. See ?tximport