Entering edit mode

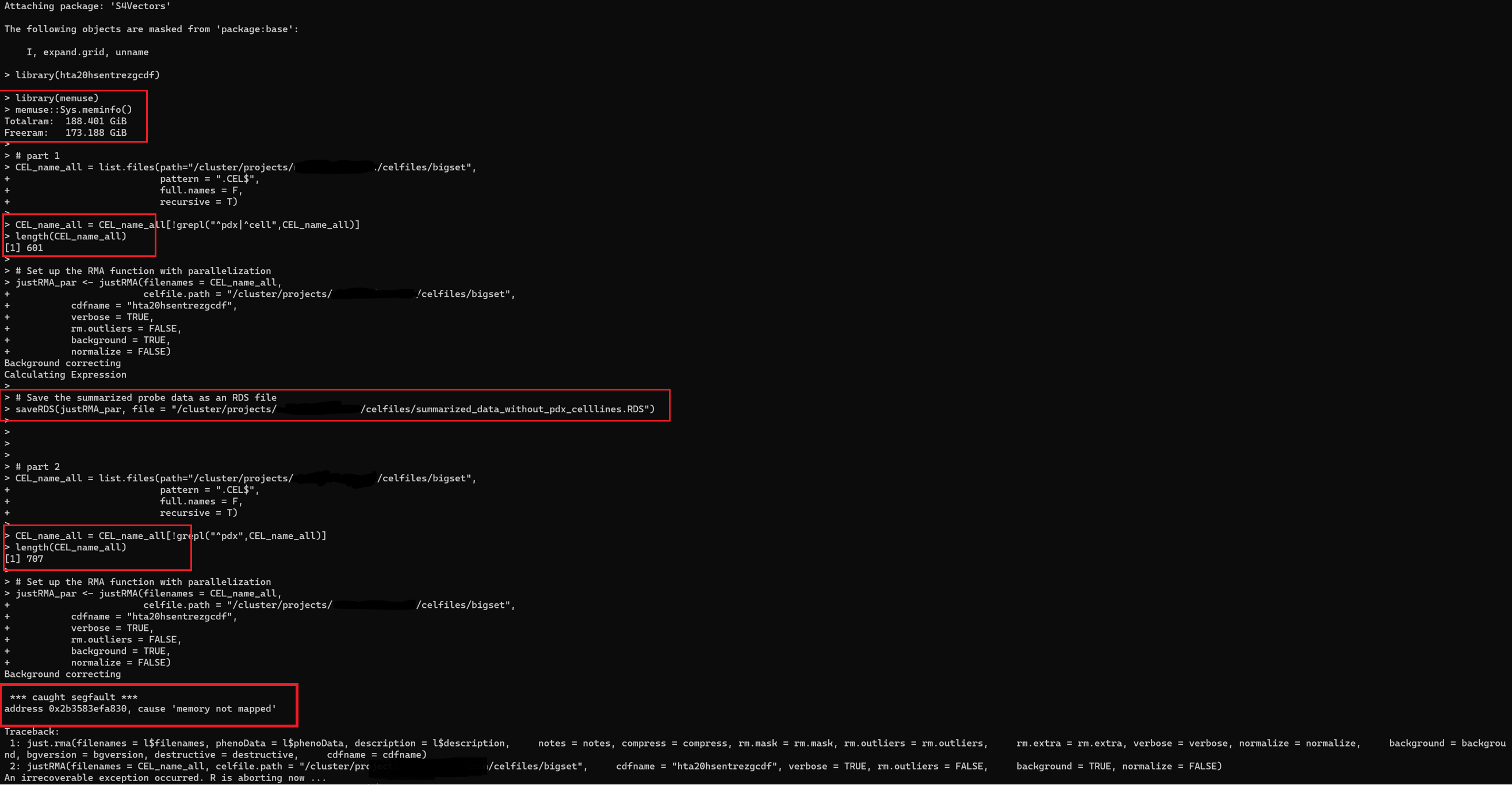
I am running affy::justRMA function on a computer cluster to make expressionset from HTA 2.0 CEL files.
The function runs fine when I run # part 1 of the code (see the attachment) , but I get the following error when I increase the number of CEL files to 700 in the # part 2 (Please see the output in the attachment)
![*** caught segfault ***
address 0x2b3583efa830, cause 'memory not mapped'][1]
Since I run this function on computer cluster I have 173GB of RAM and as you may see from the attachment R recognizes the memory ( memuse::Sys.meminfo() ).
Since the code is running with no problem with less number of samples it either the function or R isseu with memory.
- Any similar experiance? solution?
- Is it possible to use frma package for HTA CEL files with a
custom CDFfrom Brainarraylibrary(hta20hsentrezgcdf)
Best
Hossein

If you are adding a comment, please use the ADD COMMENT button, rather than ADD ANSWER.
I think you are correct - the normal error for running out of RAM is something like 'Error: a vector of length XXX could not be allocated`
This isn't an issue with
justRMA, but instead it's a C-level issue (bothrmaandjustRMAuse the same underlying C code, butjustRMAskips expensive steps like instanciating anAffyBatchfirst). The C code for theaffypackage was written years ago by Ben Bolstad, who hasn't been around these parts for years now, and people don't really use Affy arrays these days, so it's hard to get much impetus for people to want to fix an ancient codebase that works for like 99.999% of the few remaining uses.Also, the affy package was written back in the day when a given probe was only ever used for a single probeset, and for the later arrays like the HTA series Affy started sharing probes across multiple probesets. This was a problem for the affy package, and IIRC Ben Bolstad did something to patch it, but it wasn't really a priority because the
oligopackage didn't care about such things and was meant to replace affy anyway. It may be an edge case in the C code that comes up with too many arrays, but I don't know C, so am unable to be of any help for that.Also also, the
affypackage wasn't ever intended to be used for HTA arrays, and even though MBNI has remapped the probes and made a CDF package, that's really an off-label use foraffy, so I am not sure it's in anybody's interest to 'fix' a package for a use it was never intended.All that said, you might consider just processing the data in two batches. With the number of arrays you are using, I wouldn't think there would be a batch effect, and even if there were you could adjust for that in your linear model.