
Hi Michael, I am using deseq2 to perform DE analysis over ~100 human postmortem brain tissues, our major interest is finding DEGs between AD and ctrl, meanwhile we also have other covariates such as different brain regions, sexes, age, batch, and RIN (RNA integrity number). Previously I applied PCA analysis to help decide which covariates should be included in the model: design(ddssva)<- ~ SV1+SV2+Area+Sex+Sequence_Batch+Age+Group, and that did give me DEGs between AD and ctrl with functional enrichment validated as expected. But now I am a bit confused about whether the 'perfect' model design is necessary after I saw figure 2 in this paper: https://www.biologicalpsychiatryjournal.com/article/S0006-3223(20)31674-7/fulltext#secsectitle0015 According to this result from GTEx dataset, a bunch of sequencing-related technical factors appear as top drivers of expression variance for gene count matrix, which means a better model should include these technical factors as well, and that can lead to over 10 more paramters in the deseq2 model. So I have 2 questions relating to this topic:
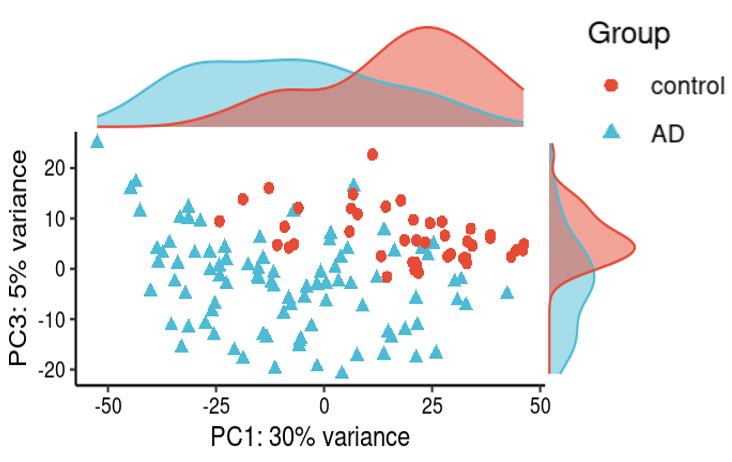
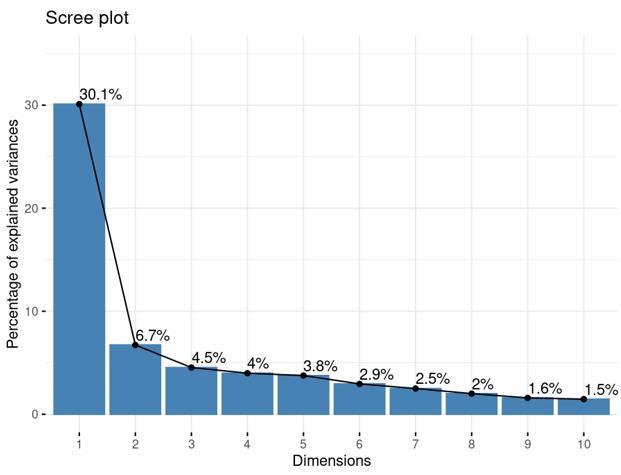
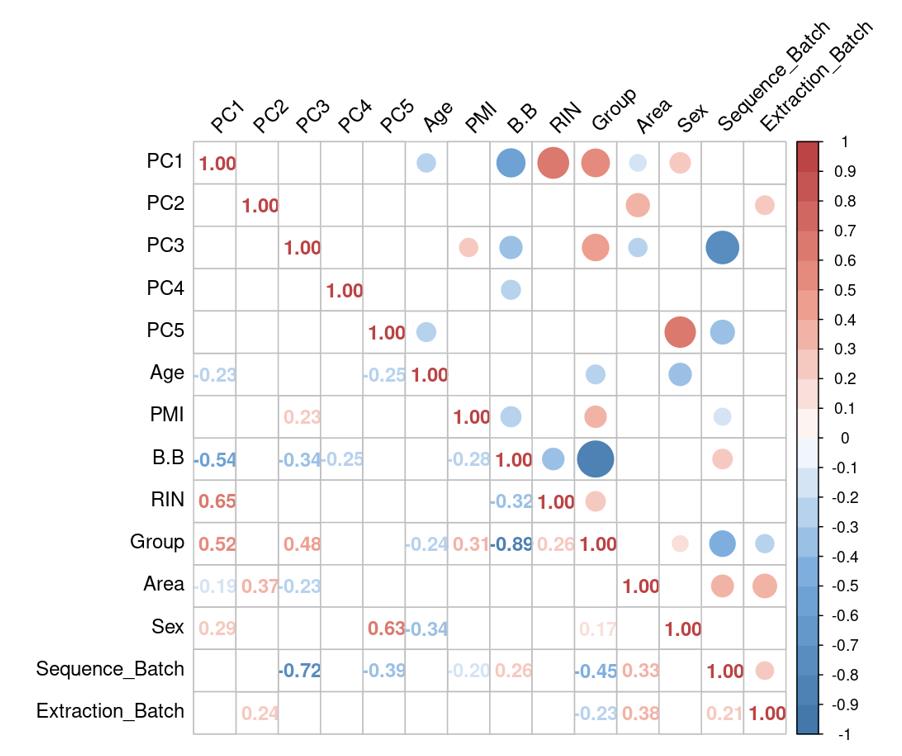
Is it reasonable to include as many covariates as possible, if those covariates are considered important judging from a top PCA plot (attached picture 1) or from the percentage of variance explained? Because on the one hand, I guess too many paramters can cause an overfitting problem; On the other hand, even if we include all those covariates, whether from the metadata sheet or sequencing fastqc report at our hand, there is still a large proportion of residual variance as you could see from figure 2 in the linked article. The similar problem also happens if we look at the PCA analysis, ideally we can have top N PCs able to explain >85% variance for the gene count matrix (in fact in some cases top N PCs can only explain some proportion of variance - attached picture 2, although we still see our expected case-ctrl signal is already there), but a correlation map (attached picture 3) between variables and top PCs shows that each variable is just strongly/weakly correlated with top N PCs. So if we use those variables in our deseq2 model, we are also not able to explain >85% variance since these variables are just partly correlated with top N PCs. This made me very confusing, is it necessary and possible to achieve a 'perfect' model design? Even though this 'perfect' model isn't perfect at all since gene count matrix has too much noise. In other words, besides the variables of interest, is there any criterion I can refer to choose which other covariates to include in the model to make my model more accurate?
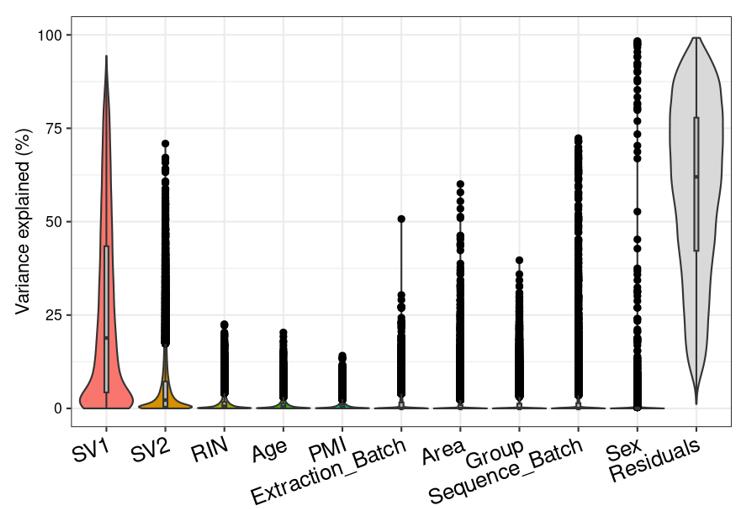
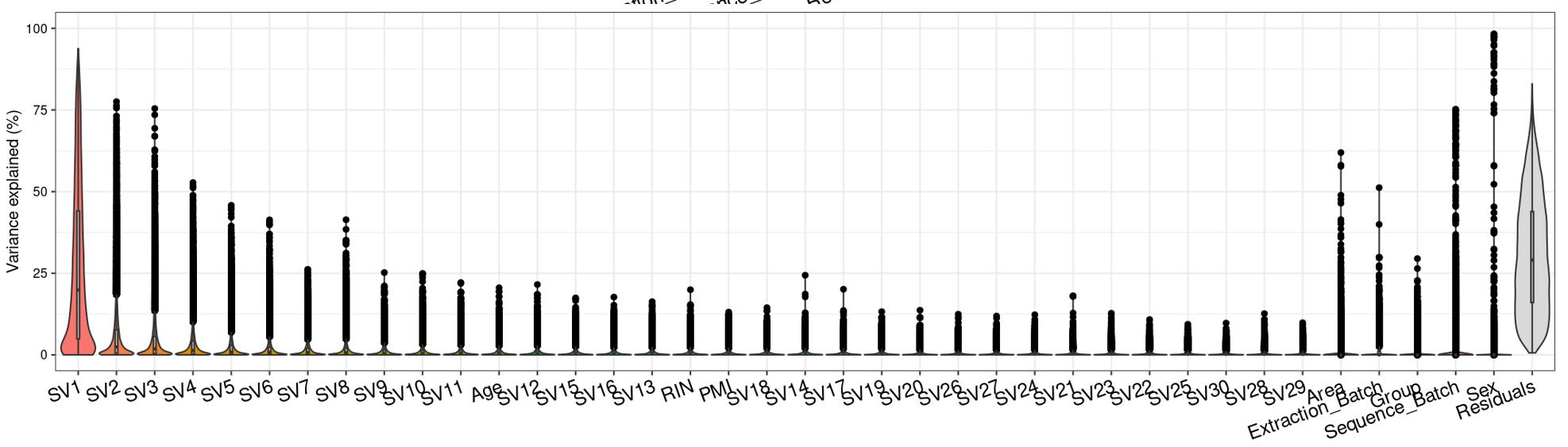
Question 2 is a extended thought of question 1, I tried using SVA (surrogate variable analysis from Rpackage: 'sva') to account for the residual influence of my current model design: design(ddssva)<- ~ SV1+SV2+Area+Sex+Sequence_Batch+Age+Group, but the same confusion happens again: how many surrogate variables should be included... Choosing top 2 SVs can give 60% residual variance (attached picture 4, variance is computed by a linear-mixed model from Rpackage: 'variancePartition', violin plot shows variance explained by each variable across all genes), while choosing top 30 SVs can give 30% residual variance (attached picture 5). But is it OK or needed that I include 30 more SVs into deseq2 model?
I really appreciate any helpful thought and ideas! Thanks a lot!


I'm not free this week but will try to return to this on Monday