
I have been troubles with using pathview command. I followed the "Pathview: pathway based data integration and visualization" and wrote the sample codes as following.
library(pathview)
data(gse16873.d)
filename=system.file("extdata/gse16873.demo",package="pathview")
gse16873=read.delim(filename,row.names=1)
gse16873.d=gse16873[,2*(1:6)]-gse16873[,2*(1:6)-1]
data(demo.paths)
data(paths.hsa)
head(paths.hsa,3)
hsa00010 hsa00020
"Glycolysis / Gluconeogenesis" "Citrate cycle (TCA cycle)"
hsa00030
"Pentose phosphate pathway"
i <- 1
pv.out <- pathview(gene.data = gse16873.d[, 1], pathway.id = demo.paths$sel.paths[i],
+ species = "hsa", out.suffix = "gse16873", kegg.native = T)
Info: Downloading xml files for hsa04110, 1/1 pathways.. Info: Downloading png files for hsa04110, 1/1 pathways.. Warning: Failed to download KEGG xml/png files, hsa04110 skipped! 경고메시지(들):
This mean for warning messages; Because the Korean R program was installed, the warning message came out in Korean. I'm sorry :(
1: download.file(xml.url, xml.target, quiet = T)에서: URL https://rest.kegg.jp/get/hsa04110/kgml: cannot open destfile './hsa04110.xml', reason 'Permission denied'
2: download.file(xml.url, xml.target, quiet = T)에서: 다운로드의 종료상태(exit status)가 0이 아닙니다.
In download.file(xml.url, xml.target, quiet = T): The exit status of the download is not 0.
I can't remember why pahtview command did not work.
My R version is following.
R version 4.3.0 alpha (2023-03-28 r84109 ucrt) Copyright (C) 2023 The R Foundation for Statistical Computing Platform: x86_64-w64-mingw32/x64 (64-bit)
I've used previous R versions (3.5.1, 4.2.2 and 4.2.3) as well, but always get stuck at this step. I'm asking because it doesn't seem to be a version issue.
Please let me know how to fix this problem.
Thank you.

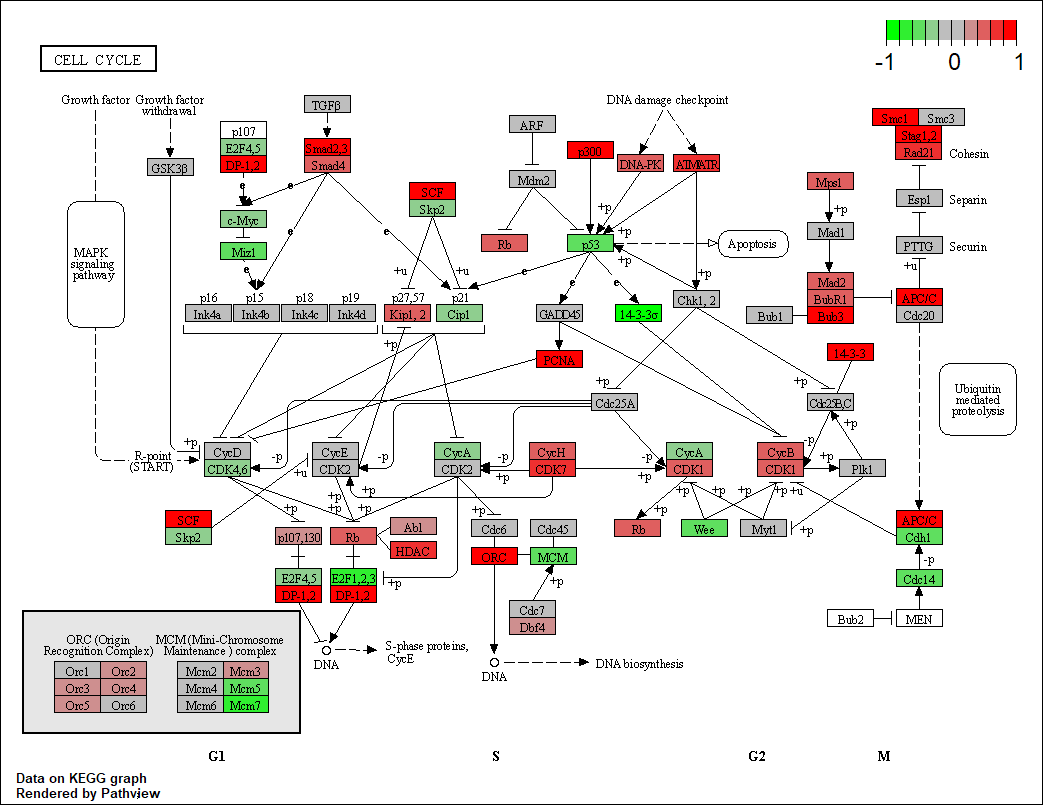
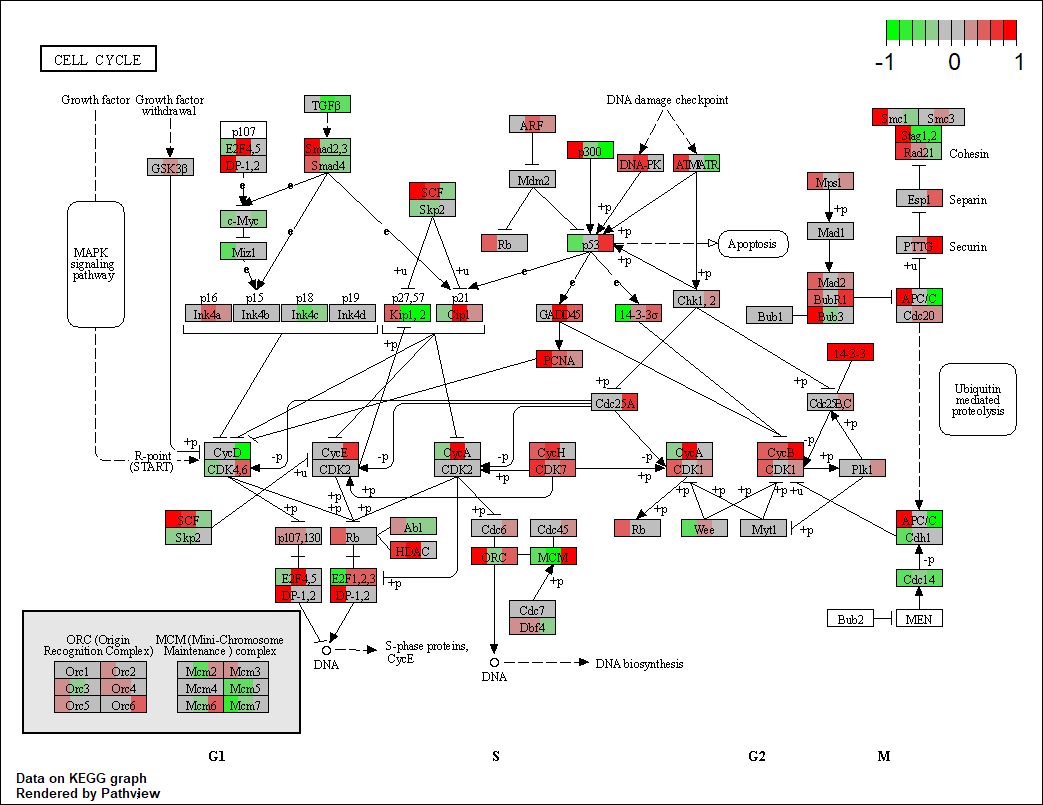
Really appreciate your help :)
At last, I can fix my problem. I have learned to check my code.