Entering edit mode

Hi, I would like to ask that the deta input fort Deseq2 is required a count metric. However, after I look into my count data, I would that there is a batch effect in my count data.
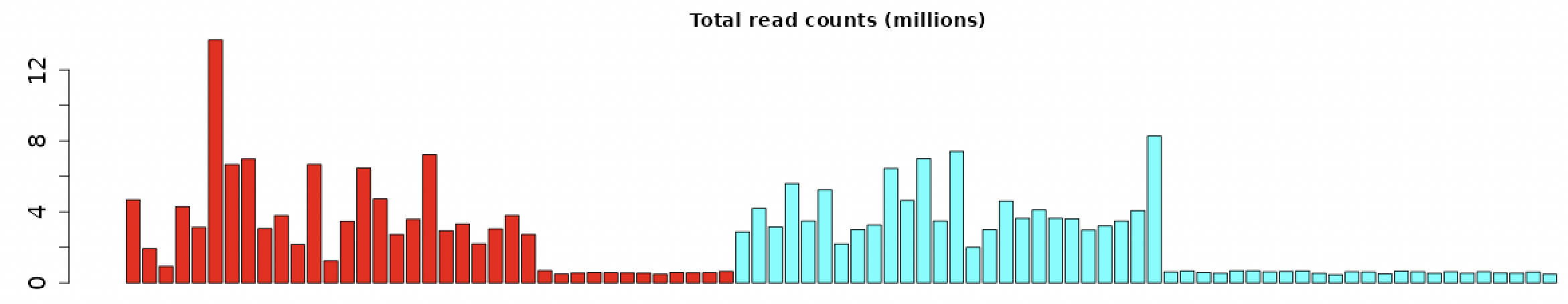
How should I normalized my read count data before applying in Deseq2 and for WGCNA analysis. Currently I have tried using Combatseq on raw read count before using in Deseq2 analysis. What is the best option in my analysis, (1) Performing batch normalization using Combatseq --> Deseq2 --> VST transformation --> WGCNA (2) DEseq2 --> VST transformation --> WGCNA
Thank you very much for your kind in advance for suggestions and comments. Code should be placed in three backticks as shown below
#define conditions, library methods, and replicates
conditions = c("N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N", "N","N", "N", "N", "N", "N", "N", "N","P","P","P", "P", "P", "P","P", "P", "P","P","P","P","P", "P", "P", "P","P", "P", "P","P","P","P","P", "P", "P", "P","P", "P", "P","P","P","P","P", "P", "P", "P","P", "P", "P","P","P","P","P", "P", "P", "P","P", "P", "P","P")
library_methods = c("D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D2","D2", "D1", "D1","D1", "D1","D1", "D1","D1", "D1","D1", "D1","D1", "D1","D2", "D2","D2", "D2","D2", "D2", "D2","D2", "D2","D2", "D2", "D2","D2", "D2","D2", "D2", "D2","D2", "D2","D2", "D2", "D2","D2", "D2","D2", "D2", "D1","D1", "D1","D1", "D1", "D1","D1", "D1","D1", "D1", "D1","D1", "D1","D1", "D1", "D1","D1", "D1","D1", "D1", "D1","D1", "D1","D1" )
replicates = c(1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25,26,1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24)
#perform the batch correction
#first we need to transform the format of our groups and batches from names (e.g. "UHR", "HBR", etc.) to numbers (e.g. 1, 2, etc.)
#in the command below "sapply" is used to apply the "switch" command to each element and convert names to numbers as we define
groups = sapply(as.character(conditions), switch, "N" = 1, "P" = 2, USE.NAMES = F)
batches = sapply(as.character(library_methods), switch, "D1" = 1, "D2" = 2, USE.NAMES = F)
sample_names = names(uncorrected_data)[2:length(names(uncorrected_data))]
#now run ComBat_seq
corrected_data = ComBat_seq(counts = as.matrix(uncorrected_data[,sample_names]), batch = batches, group = groups)
#join the gene and chromosome names onto the now corrected counts from ComBat_seq
#corrected_data = cbind(uncorrected_data[,c("gene", "chr")], corrected_data)
corrected_data = cbind(uncorrected_data[,c("gene")], corrected_data)
# include your problematic code here with any corresponding output
# please also include the results of running the following in an R session
sessionInfo( )

Hi, thank you very much for your answer.
As I have seen some articles about normalization, I have a confusion a little bit about the different between quartile normalization and the normalization in Deseq2.
Regarding to my result of total count, we would observe that there are differences in total count in each sample. from the figure in the post above, we can see that the batch 2 shown slightly high in total count value.
In this case, how should I normalize all the read count before differential gene expression analysis and WGCNA?
Use the default normalization in DESeq2, nothing special here.