
Hi everyone,
I am looking at one of the ImmGen RNASeq dataset and I am looking at a specific subset of genes, corresponding to a family of interest.
The first step I performed was to subset the counts for the genes that I wanted (~1357 genes) and then plot the PCA for the samples for that (I had to add a pseudocount in order for it to run). The results were the following (I faceted them by organ to be "cleaner" to see any possible pattern in other subgroups):
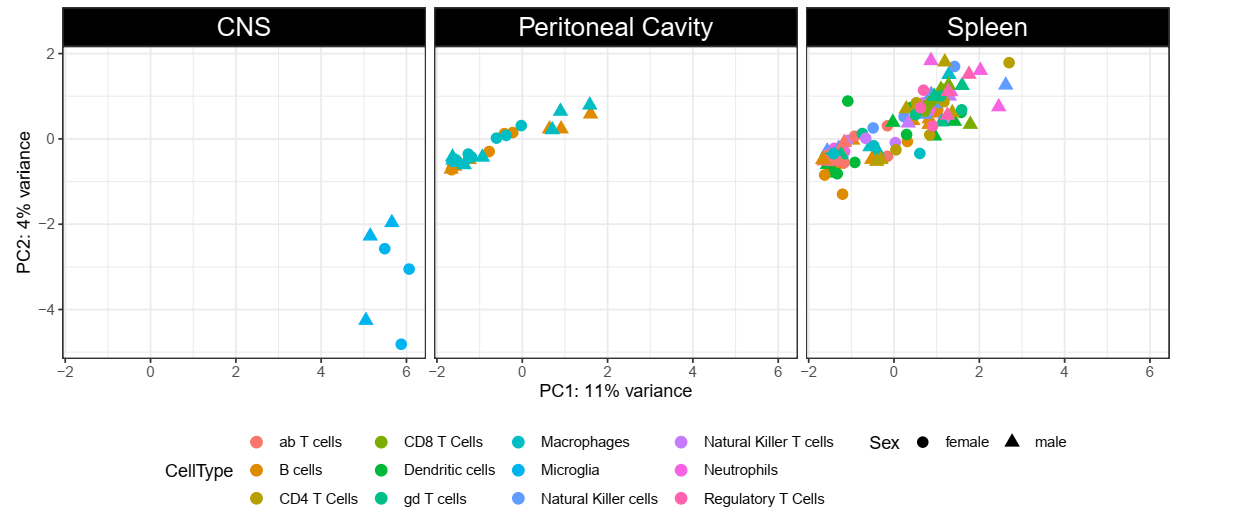
However, when performing DE in the full dataset (without any prior filtering), I get a lot of differences in the subset genes of interest between two groups that are appearing together in the clustering (Spleen and Peritoneal Cavity):
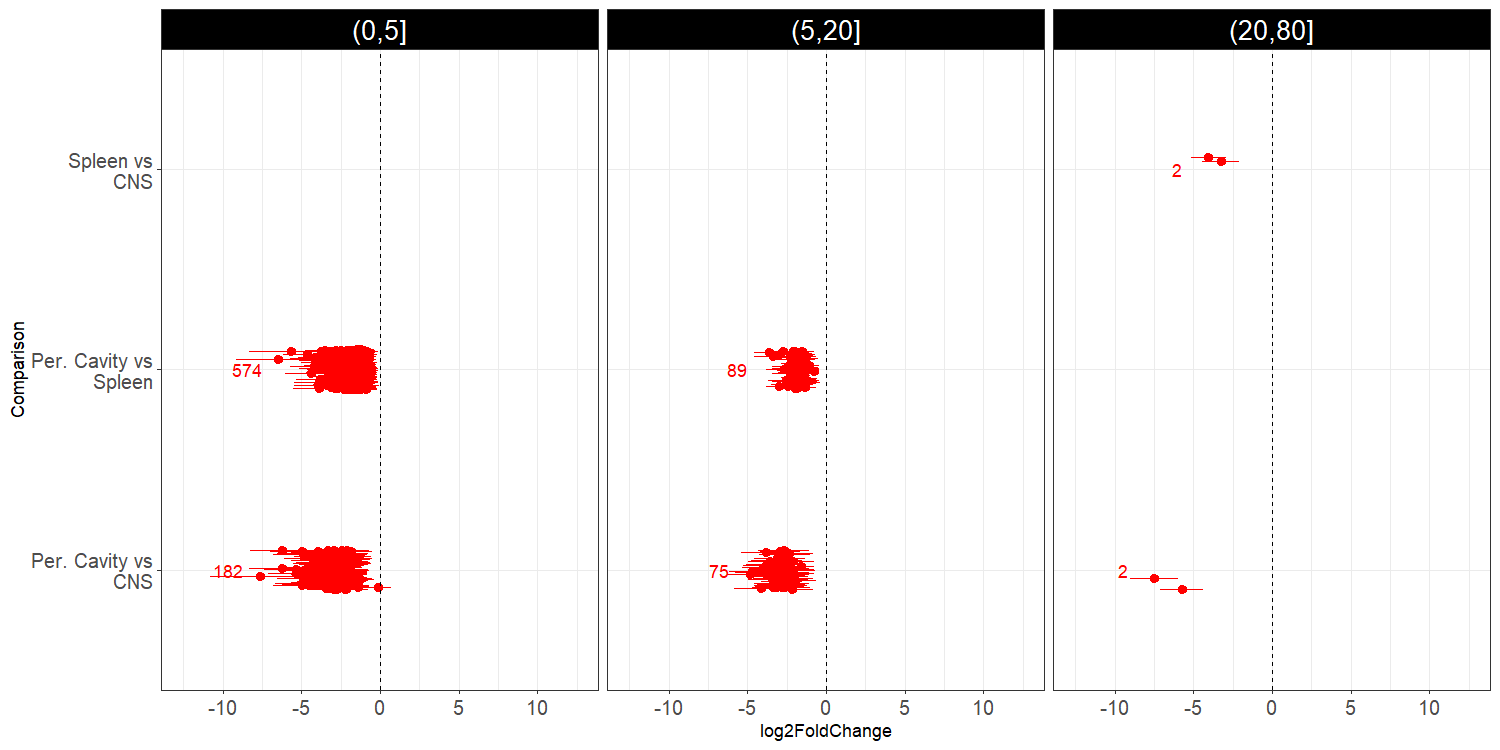
I faceted these results by the value of the highest base mean per group from the two groups in the pair-wise comparisons. What I really wanted to know is if this makes sense to you. I fear that, by subsetting the data for the PCA I might be causing some artifacts in the clustering.
Thanks in advance.
EDIT: I added some information that was relevant to understanding my problem, specifically the number of genes of interest and that the DE results are also for that subset (performed the DE for the whole dataset but, then filtered the results for the genes of interest).

Hey! Thanks for your answer. I just added some information that was missing that could improve the understanding of my problem. The DE results are filtered for the subset of genes of interest (despite the analysis being ran for the full dataset).
Thanks!