
Hello, im struggling to load my data into topGo. I have been able to load in the preinstalled data but when i try my own i get the error
Error in split.default(names(sort(nl)), f.index): first argument must be a vector
I have found some similar post/problems but i still cannot identify the issue.
Im bringing over gene lists and p values from a DESEQ2 output and loading that as a genelist, along with a GO file for my organism as follows;
# extract the adjusted p-values from our results object
geneList <- res$padj
# add the gene names from our results to our list of adjusted p-values
names(geneList) <- rownames(res)
# remove genes with an adjusted p-value recorded as NA
geneList<-na.omit(geneList)
#load in GO data from B.imp
geneID2GO <- readMappings(file = "Bombus_impatiens_BP_All.txt", IDsep = ",", sep = "\t")
sessionInfo( )
these data look like;
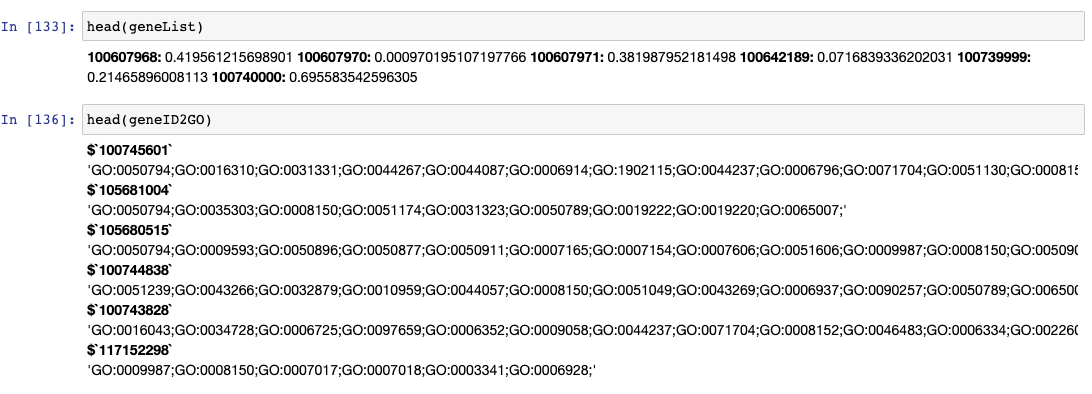
but when i run topGo, i get the error
sampleGOdata <- new("topGOdata",
ontology = "BP",
allGenes = geneList,
geneSel = topDiffGenes,
nodeSize = 10,
annot = annFUN.gene2GO,
gene2GO = geneID2GO
)
Building most specific GOs .....
( 0 GO terms found. )
Build GO DAG topology ..........
( 0 GO terms and 0 relations. )
Nothing to do:
Error in split.default(names(sort(nl)), f.index): first argument must be a vector
Traceback:
1. new("topGOdata", ontology = "BP", allGenes = geneList, geneSel = topDiffGenes,
. nodeSize = 10, annot = annFUN.gene2GO, gene2GO = geneID2GO)
2. initialize(value, ...)
3. initialize(value, ...)
4. .local(.Object, ...)
5. buildLevels(g, leafs2root = TRUE)
6. split(names(sort(nl)), f.index)
7. split.default(names(sort(nl)), f.index)
Based on other posts i thought maybe my genelist file wasnt a vector but;
typeof(geneList)
'double'
And if i run
v.geneList <- as.vector(geneList,'numeric')
sessionInfo( )
and put that through, i still get the error.
Could it be the geneID2GO file? the format looks like it should so im pretty stumped! Any suggestions appreciated!