
Hello everyone! I'm new in RNAseq data analysis and trying to apply a pipeline from an online course to A.thaliana data in my university course project. The pipeline includes kallisto quantification with subsequent analysis in R. I performed kallisto quantification with A.thaliana transcriptome, which I downloaded from plants.ensembl.org, got all the esential files. Firstly, I imported studydesign and set paths to abundance.tsv files:
targets <- read_tsv("ATstudydesign.txt")
path <- file.path(targets$Sample[1:12], "abundance.tsv")
Now, the main trouble is how to import data from abundance.tsv files to R project and to replace transcript names with gene names.
I need a tx2gene object for tximport() function and, as I understand, there are several ways to create it.
I tried to create it by downloading a table from https://plants.ensembl.org/biomart/martview/18f83aa36cd9df8402183dba5ee6d603
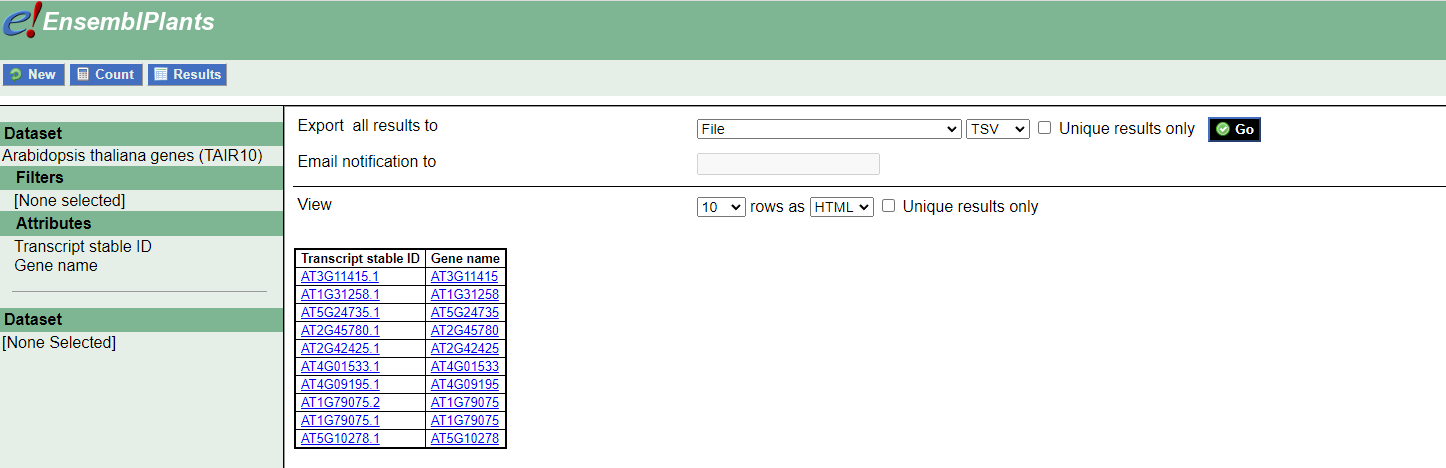
Then I imported it into R project:
Tx <- read_tsv("mart_export.txt")
head(Tx)
The Tx file looks good since it has transcripts names in the first column and gene names in the second:
> head(Tx)
# A tibble: 6 × 2
`Transcript stable ID` `Gene name`
<chr> <chr>
1 AT3G11415.1 AT3G11415
2 AT1G31258.1 AT1G31258
3 AT5G24735.1 AT5G24735
4 AT2G45780.1 AT2G45780
5 AT2G42425.1 AT2G42425
6 AT4G01533.1 AT4G01533
I ran tximport() function but got an error:
> Txi_gene <- tximport(path,
+ type = "kallisto",
+ tx2gene = Tx,
+ txOut = FALSE,
+ countsFromAbundance = "lengthScaledTPM",
+ ignoreTxVersion = TRUE)
Note: importing `abundance.h5` is typically faster than `abundance.tsv`
reading in files with read_tsv
1 2 3 4 5 6 7 8 9 10 11 12
Error in .local(object, ...) :
None of the transcripts in the quantification files are present
in the first column of tx2gene. Check to see that you are using
the same annotation for both.
Example IDs (file): [AT1G76820, ATMG00060, AT4G16360, ...]
Example IDs (tx2gene): [AT3G11415.1, AT1G31258.1, AT5G24735.1, ...]
This can sometimes (not always) be fixed using 'ignoreTxVersion' or 'ignoreAfterBar'.
I tried to look at abundance.tsv file by importing the data from it:
Sample <- read_tsv("./RootCtrl1/abundance.tsv")
> head(Sample)
# A tibble: 6 × 5
target_id length eff_length est_counts tpm
<chr> <dbl> <dbl> <dbl> <dbl>
1 AT1G76820.1 3857 3645. 21.8 0.934
2 ATMG00060.1 542 333. 2 0.939
3 AT4G16360.1 1685 1473. 465 49.3
4 AT5G26800.1 637 426. 166 60.9
5 AT4G16110.1 2666 2454. 293 18.7
6 AT5G39100.1 808 597. 0 0
I also tried to search several transcript names in abundance.tsv and mart_export.txt and I was able to find them in both files through simple Find in Sublime text editor.
Can't understand what's wrong with these commands and/or files. Maybe someone will share thoughts or suuggestions? Maybe there are alternative ways to create this tx2gene objects? Thanks in advance!
sessionInfo( )
> sessionInfo()
R version 4.2.2 (2022-10-31 ucrt)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19044)
Matrix products: default
locale:
[1] LC_COLLATE=Russian_Russia.utf8 LC_CTYPE=Russian_Russia.utf8 LC_MONETARY=Russian_Russia.utf8
[4] LC_NUMERIC=C LC_TIME=Russian_Russia.utf8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] tximport_1.26.0 forcats_0.5.2 stringr_1.4.1 dplyr_1.0.10 purrr_0.3.5 readr_2.1.3 tidyr_1.2.1
[8] tibble_3.1.8 ggplot2_3.4.0 tidyverse_1.3.2
loaded via a namespace (and not attached):
[1] bitops_1.0-7 matrixStats_0.62.0 fs_1.5.2 lubridate_1.9.0
[5] bit64_4.0.5 filelock_1.0.2 progress_1.2.2 httr_1.4.4
[9] GenomeInfoDb_1.32.2 tools_4.2.2 backports_1.4.1 utf8_1.2.2
[13] R6_2.5.1 DBI_1.1.3 BiocGenerics_0.42.0 colorspace_2.0-3
[17] rhdf5filters_1.8.0 withr_2.5.0 tidyselect_1.2.0 prettyunits_1.1.1
[21] bit_4.0.4 curl_4.3.3 compiler_4.2.2 rvest_1.0.3
[25] cli_3.3.0 Biobase_2.56.0 xml2_1.3.3 DelayedArray_0.22.0
[29] rtracklayer_1.56.1 scales_1.2.1 rappdirs_0.3.3 digest_0.6.30
[33] Rsamtools_2.12.0 XVector_0.36.0 pkgconfig_2.0.3 MatrixGenerics_1.8.1
[37] dbplyr_2.2.1 fastmap_1.1.0 readxl_1.4.1 rlang_1.0.6
[41] rstudioapi_0.14 RSQLite_2.2.15 BiocIO_1.6.0 generics_0.1.3
[45] jsonlite_1.8.3 vroom_1.6.0 BiocParallel_1.30.3 googlesheets4_1.0.1
[49] RCurl_1.98-1.8 magrittr_2.0.3 GenomeInfoDbData_1.2.8 Matrix_1.5-1
[53] Rhdf5lib_1.18.2 Rcpp_1.0.9 munsell_0.5.0 S4Vectors_0.34.0
[57] fansi_1.0.3 lifecycle_1.0.3 stringi_1.7.8 yaml_2.3.6
[61] SummarizedExperiment_1.26.1 zlibbioc_1.42.0 rhdf5_2.40.0 BiocFileCache_2.4.0
[65] grid_4.2.2 blob_1.2.3 parallel_4.2.2 crayon_1.5.2
[69] lattice_0.20-45 Biostrings_2.64.0 haven_2.5.1 GenomicFeatures_1.48.3
[73] hms_1.1.2 KEGGREST_1.36.3 pillar_1.8.1 GenomicRanges_1.48.0
[77] rjson_0.2.21 codetools_0.2-18 biomaRt_2.52.0 stats4_4.2.2
[81] reprex_2.0.2 XML_3.99-0.10 glue_1.6.2 modelr_0.1.10
[85] tzdb_0.3.0 png_0.1-7 vctrs_0.5.0 cellranger_1.1.0
[89] gtable_0.3.1 assertthat_0.2.1 cachem_1.0.6 broom_1.0.1
[93] restfulr_0.0.15 googledrive_2.0.0 gargle_1.2.1 GenomicAlignments_1.32.1
[97] AnnotationDbi_1.58.0 memoise_2.0.1 IRanges_2.30.0 timechange_0.1.1
[101] ellipsis_0.3.2

Thank you for your answer! Of course I read the error message before posting and tried to switch
ignoreTxVersionback and forth. It seems to me that whenTRUEit ignores versions of transcripts only in abundance.tsv files, not in tx2gene. I tried another way to create tx2gene table with GenomicFeatures package:txdb <- makeTxDbFromBiomart(biomart = "plants_mart", host="https://plants.ensembl.org", dataset = "athaliana_eg_gene")Then I got the same error and deleted versions from transcripts. The final version isI used advices from https://sbc.shef.ac.uk/workshops/2020-02-13-rnaseq-r/rna-seq-preprocessing.nb.html
I think the code can be optimised but it works:)
Ah, yes. The exact opposite issue for which ignoreTxVersion is intended.
Your solution obviously works, but as you note could be optimized. The tidyverse IMO mostly re-invents existing wheels while requiring more typing to do so. You can do the exact same thing with just three lines of code, which is optimized in terms of typing, and likely in terms of speed as well.